
Computational and Mass Spectrometry-Based Approach Identify Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) in JMJD6


Abstract
:1. Introduction
2. Results
2.1. Deleterious nsSNPs Predicted in JMJD6
2.2. Inferences of Damaging Effect of Disease-Related nsSNPs Using Multiple Approaches
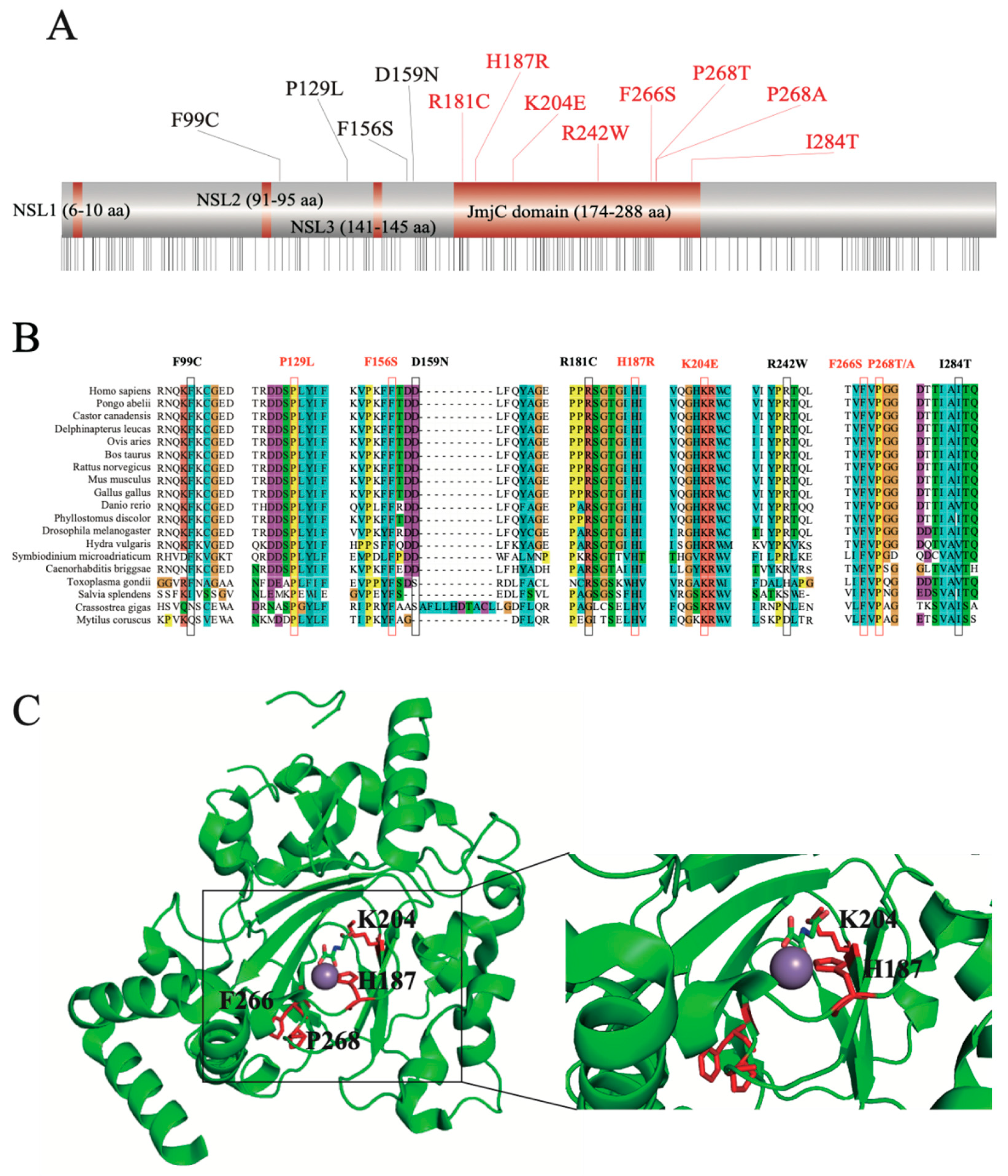
2.3. Structural Feature-Based Functional Analysis Nominated Crucial nsSNPs
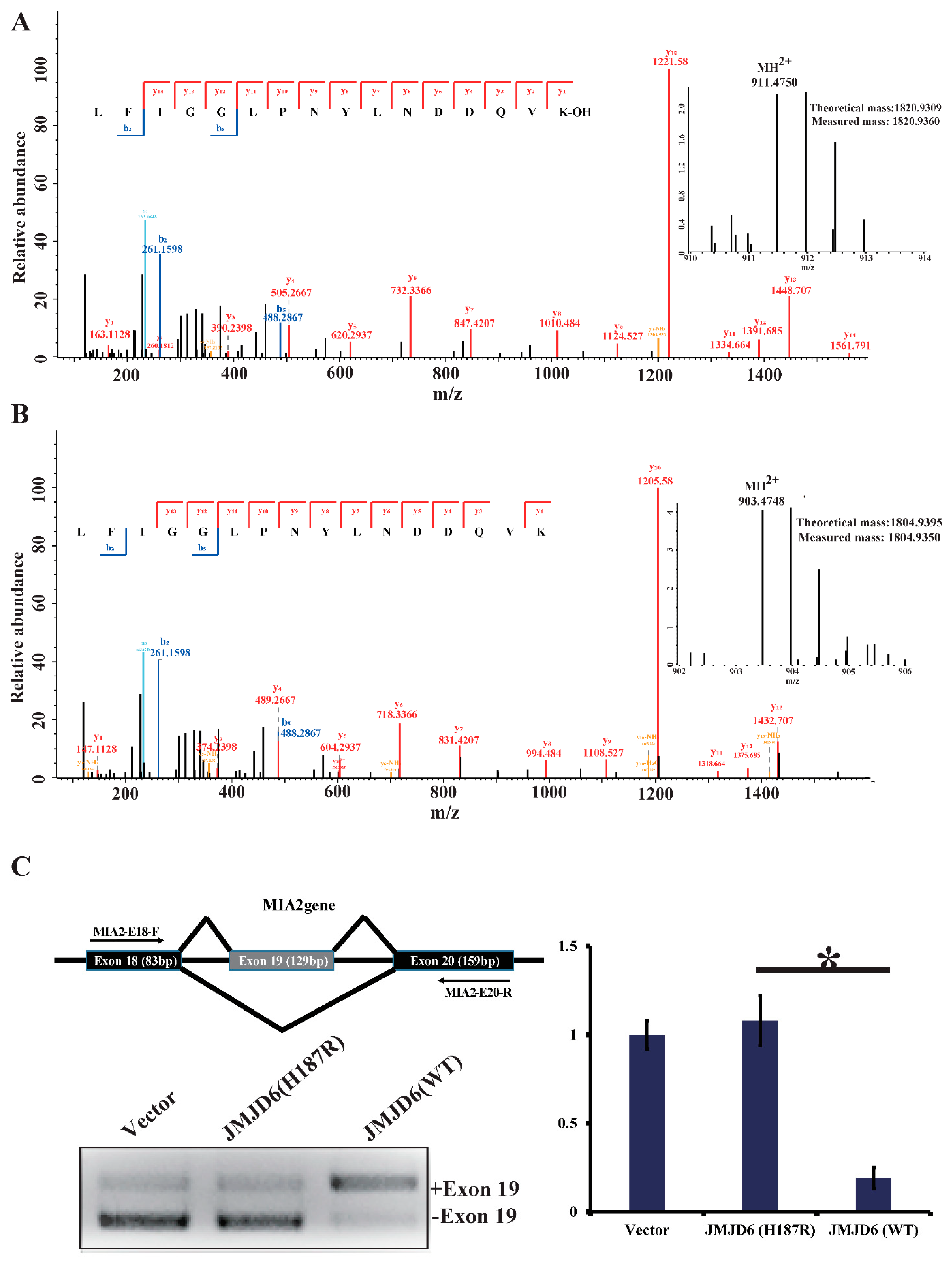
2.4. JMJD6 (H187R) Abolishes Lysyl-Hydroxylation to U2AF65 and Influences mRNA Splicing
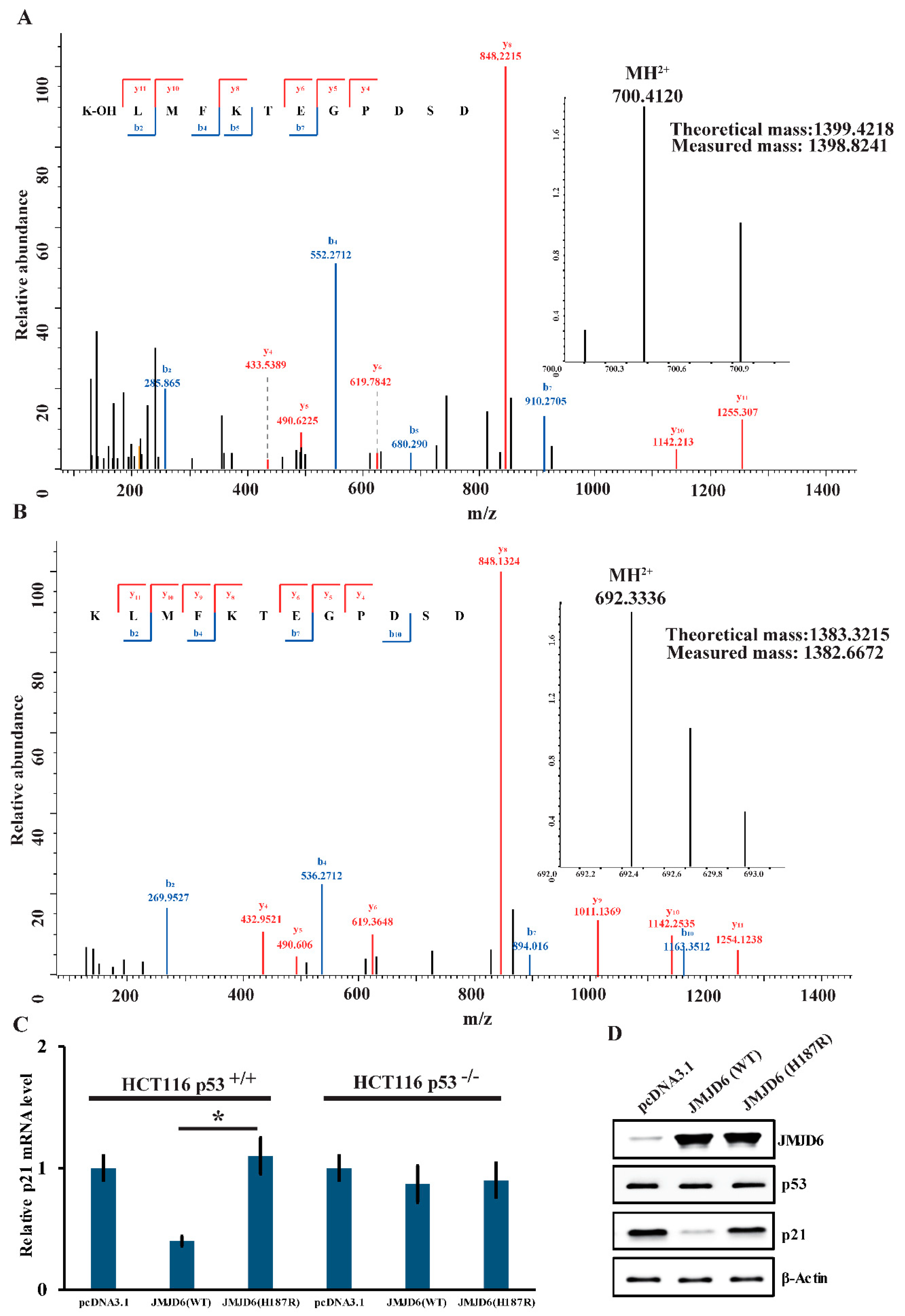
2.5. JMJD6 (H187R) Abolishes Lysyl-Hydroxylation to p53 and Can’t Inhibit p21 Expression
3. Discussion
4. Materials and Methods
4.1. Data Mining
4.2. Prediction of Functional Consequences of Non-Synonymous Coding SNPs
4.3. Sequence Homology-Based Prediction of Damaging nsSNPs by SIFT
4.4. Structure Homology-Based Prediction of Damaging nsSNPs by PolyPhen-2
4.5. Functional Consequences Prediction Based on Neural Network Classification by SNAP
4.6. Protein Stability Changes Predicted by I-Mutant 2.0
4.7. Disease-Related Prediction of nsSNPs by PhD-SNP, SNPs&GO, and PANTHER
4.8. Plasmids, Antibodies, and Reagents
4.9. Cell Culture and Transfection
4.10. Hydroxylation Assay
4.11. SDS-PAGE and In-Gel Digestion
4.12. Western Blot
4.13. Real-Time Reverse Transcription PCR
4.14. Protein Identification and Quantification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Arenas, C.; Caceres, A.; Moreno, V.; Gonzalez, J.R. Common polymorphic inversions at 17q21.31 and 8p23.1 associate with cancer prognosis. Hum. Genom. 2019, 13, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.; Sun, Y.; Xi, Z.; Milazzo, G.; Poulos, R.C.; Bartenhagen, C.; Bell, J.L.; Mayoh, C.; Ho, N.; Tee, A.E.; et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 2019, 10, 3319. [Google Scholar] [CrossRef] [Green Version]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, S.; Yang, Y.; Ma, X.; Shao, B.; Yang, S.; Wei, Y.; Wei, X. Jumonji domain-containing protein 6 protein and its role in cancer. Cell Prolif. 2020, 53, e12747. [Google Scholar] [CrossRef] [Green Version]
- Poulard, C.; Rambaud, J.; Lavergne, E.; Jacquemetton, J.; Renoir, J.M.; Tredan, O.; Chabaud, S.; Treilleux, I.; Corbo, L.; Le Romancer, M. Role of JMJD6 in Breast Tumourigenesis. PLoS ONE 2015, 10, e0126181. [Google Scholar] [CrossRef]
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R., 3rd; et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009, 325, 90–93. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zhen, Z.; Qi, H.; Yuan, X.; Gao, X.; Zhang, M. U2AF65 enhances milk synthesis and growth of bovine mammary epithelial cells by positively regulating the mTOR-SREBP-1c signalling pathway. Cell Biochem. Funct. 2019, 37, 93–101. [Google Scholar] [CrossRef]
- Yates, C.M.; Sternberg, M.J. The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein-protein interactions. J. Mol. Biol. 2013, 425, 3949–3963. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genom. 2014, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef] [Green Version]
- Roach, J.C.; Glusman, G.; Smit, A.F.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef] [Green Version]
- Dakal, T.C.; Kala, D.; Dhiman, G.; Yadav, V.; Krokhotin, A.; Dokholyan, N.V. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms in IL8 gene. Sci. Rep. 2017, 7, 6525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamali, Z.; Taheri-Anganeh, M.; Entezam, M. Prediction of potential deleterious nonsynonymous single nucleotide polymorphisms of HIF1A gene: A computational approach. Comput. Biol. Chem. 2020, 88, 107354. [Google Scholar] [CrossRef]
- Chitrala, K.N.; Nagarkatti, M.; Nagarkatti, P.; Yeguvapalli, S. Analysis of the TP53 Deleterious Single Nucleotide Polymorphisms Impact on Estrogen Receptor Alpha-p53 Interaction: A Machine Learning Approach. Int. J. Mol. Sci. 2019, 20, 2962. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.; Kulak, N.A.; Nagaraj, N.; Cox, J. The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 2013, 49, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaro, J.A.; Ignatchenko, A.; Ignatchenko, V.; Sinha, A.; Boutros, P.C.; Kislinger, T. Detecting protein variants by mass spectrometry: A comprehensive study in cancer cell-lines. Genome Med. 2017, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, T.; Nishiyama, R.; Shimogori, T.; Nukina, N. Proteomics-Based Approach Identifies Altered ER Domain Properties by ALS-Linked VAPB Mutation. Sci. Rep. 2020, 10, 7610. [Google Scholar] [CrossRef] [PubMed]
- Schessl, J.; Zou, Y.; McGrath, M.J.; Cowling, B.S.; Maiti, B.; Chin, S.S.; Sewry, C.; Battini, R.; Hu, Y.; Cottle, D.L.; et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J. Clin. Investig. 2008, 118, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Chen, Y.; Liu, J.; Xu, L.; Zhang, Z.; Zhang, J.; Wang, H.; Wang, X.; Chen, J.; Zhao, R.; et al. Proteomics screen to reveal molecular changes mediated by C722G missense mutation in CHRM2 gene. J. Proteom. 2013, 89, 39–50. [Google Scholar] [CrossRef]
- Nishimura, T.; Vegvari, A.; Nakamura, H.; Kato, H.; Saji, H. Mutant Proteomics of Lung Adenocarcinomas Harboring Different EGFR Mutations. Front. Oncol. 2020, 10, 1494. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, S.K.; Cavey, G.S.; Pearson, J.D. Single ribosomal protein mutations in antibiotic-resistant bacteria analyzed by mass spectrometry. Antimicrob. Agents Chemother. 2001, 45, 3046–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, A.L.; Eckhardt, M.; Krogan, N.J. Mass spectrometry-based protein-protein interaction networks for the study of human diseases. Mol. Syst. Biol. 2021, 17, e8792. [Google Scholar] [CrossRef]
- Hahn, P.; Bose, J.; Edler, S.; Lengeling, A. Genomic structure and expression of Jmjd6 and evolutionary analysis in the context of related JmjC domain containing proteins. BMC Genom. 2008, 9, 293. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; He, L.; Huangyang, P.; Liang, J.; Si, W.; Yan, R.; Han, X.; Liu, S.; Gui, B.; Li, W.; et al. JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS Biol. 2014, 12, e1001819. [Google Scholar] [CrossRef] [Green Version]
- Sickmier, E.A.; Frato, K.E.; Shen, H.; Paranawithana, S.R.; Green, M.R.; Kielkopf, C.L. Structural basis for polypyrimidine tract recognition by the essential pre-mRNA splicing factor U2AF65. Mol. Cell 2006, 23, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Hastings, M.L.; Allemand, E.; Duelli, D.M.; Myers, M.P.; Krainer, A.R. Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF(65). PLoS ONE 2007, 2, e538. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends. Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Singh, A.; Thakur, M.; Singh, S.K.; Sharma, L.K.; Chandra, K. Exploring the effect of nsSNPs in human YPEL3 gene in cellular senescence. Sci. Rep. 2020, 10, 15301. [Google Scholar] [CrossRef] [PubMed]
- Ait El Cadi, C.; Krami, A.M.; Charoute, H.; Elkarhat, Z.; Sifeddine, N.; Lakhiari, H.; Rouba, H.; Barakat, A.; Nahili, H. Prediction of the Impact of Deleterious Nonsynonymous Single Nucleotide Polymorphisms on the Human RRM2B Gene: A Molecular Modeling Study. Biomed. Res. Int. 2020, 2020, 7614634. [Google Scholar] [CrossRef]
- Yazar, M.; Ozbek, P. In Silico Tools and Approaches for the Prediction of Functional and Structural Effects of Single-Nucleotide Polymorphisms on Proteins: An Expert Review. OMICS J. Integr. Biol. 2021, 25, 23–37. [Google Scholar] [CrossRef]
- Hasnain, M.J.U.; Shoaib, M.; Qadri, S.; Afzal, B.; Anwar, T.; Abbas, S.H.; Sarwar, A.; Talha Malik, H.M.; Tariq Pervez, M. Computational analysis of functional single nucleotide polymorphisms associated with SLC26A4 gene. PLoS ONE 2020, 15, e0225368. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakamura, H.; Vegvari, A.; Marko-Varga, G.; Furuya, N.; Saji, H. Current status of clinical proteogenomics in lung cancer. Expert Rev. Proteom. 2019, 16, 761–772. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R. WS-SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genom. 2013, 14 (Suppl. 3), S6. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP ID | Amino Acid Change | SIFT | PolyPhen | SNAP | I-MUTANT | PhD-SNP | PANTHER | SNPs&GO |
|---|---|---|---|---|---|---|---|---|
| rs770686748 | R411W | D | PD | D | D | N | — | N |
| rs759427088 | G409E | D | PD | D | D | N | N | N |
| rs769402176 | R399H | D | PD | D | D | N | N | N |
| rs1177861863 | D393H | D | PD | D | D | N | N | N |
| rs751792177 | R373H | D | PD | D | D | D | D | N |
| rs757164575 | S352C | D | PD | D | D | N | D | N |
| rs1157910263 | S340C | D | PD | D | D | N | D | N |
| rs1417542107 | I284T | D | PD | D | D | D | D | D |
| rs778790592 | P268T | D | PD | D | D | D | D | D |
| rs778790592 | P268A | D | PD | D | D | D | D | D |
| rs1418743067 | F266S | D | PD | D | D | D | D | D |
| rs374399276 | R242W | D | PD | D | D | D | D | D |
| rs765486191 | R205H | D | PD | D | D | D | N | N |
| rs766654214 | R205C | D | PD | D | D | D | D | N |
| rs758184469 | K204E | D | PD | D | D | D | D | D |
| rs1159480887 | H187R | D | PD | D | D | D | D | D |
| rs1366225731 | R181C | D | PD | D | D | D | D | D |
| rs1162409498 | D159N | D | PD | D | D | D | D | D |
| rs1381511354 | F156S | D | PD | D | D | D | D | D |
| rs748652403 | D149V | D | PD | D | D | N | N | N |
| rs369981508 | P129L | D | PD | D | D | D | D | D |
| rs1301580761 | D126G | D | PD | D | D | D | N | N |
| rs1394232718 | Y117H | D | PD | D | D | D | D | N |
| rs1398491957 | F99C | D | PD | D | D | D | D | D |
| rs746020005 | Y94C | D | PD | D | D | D | N | N |
| rs750848447 | R28W | D | PD | D | D | D | — | N |
| rs1490052400 | S23W | D | PD | D | D | D | — | N |
| rs1278674934 | R8C | D | PD | D | D | N | — | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, T.; Yang, L.; Shen, F.; Chen, H.; Pan, Z.; Zhang, Q.; Jiang, Y.; Zhong, F.; Yang, P.; Zhang, Y. Computational and Mass Spectrometry-Based Approach Identify Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) in JMJD6. Molecules 2021, 26, 4653. https://doi.org/10.3390/molecules26154653
Gong T, Yang L, Shen F, Chen H, Pan Z, Zhang Q, Jiang Y, Zhong F, Yang P, Zhang Y. Computational and Mass Spectrometry-Based Approach Identify Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) in JMJD6. Molecules. 2021; 26(15):4653. https://doi.org/10.3390/molecules26154653
Chicago/Turabian StyleGong, Tianqi, Lujie Yang, Fenglin Shen, Hao Chen, Ziyue Pan, Quanqing Zhang, Yan Jiang, Fan Zhong, Pengyuan Yang, and Yang Zhang. 2021. "Computational and Mass Spectrometry-Based Approach Identify Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) in JMJD6" Molecules 26, no. 15: 4653. https://doi.org/10.3390/molecules26154653
APA StyleGong, T., Yang, L., Shen, F., Chen, H., Pan, Z., Zhang, Q., Jiang, Y., Zhong, F., Yang, P., & Zhang, Y. (2021). Computational and Mass Spectrometry-Based Approach Identify Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) in JMJD6. Molecules, 26(15), 4653. https://doi.org/10.3390/molecules26154653