
Detection of 4a,5-dihydropravastatin as Impurity in the Cholesterol Lowering Drug Pravastatin



Abstract
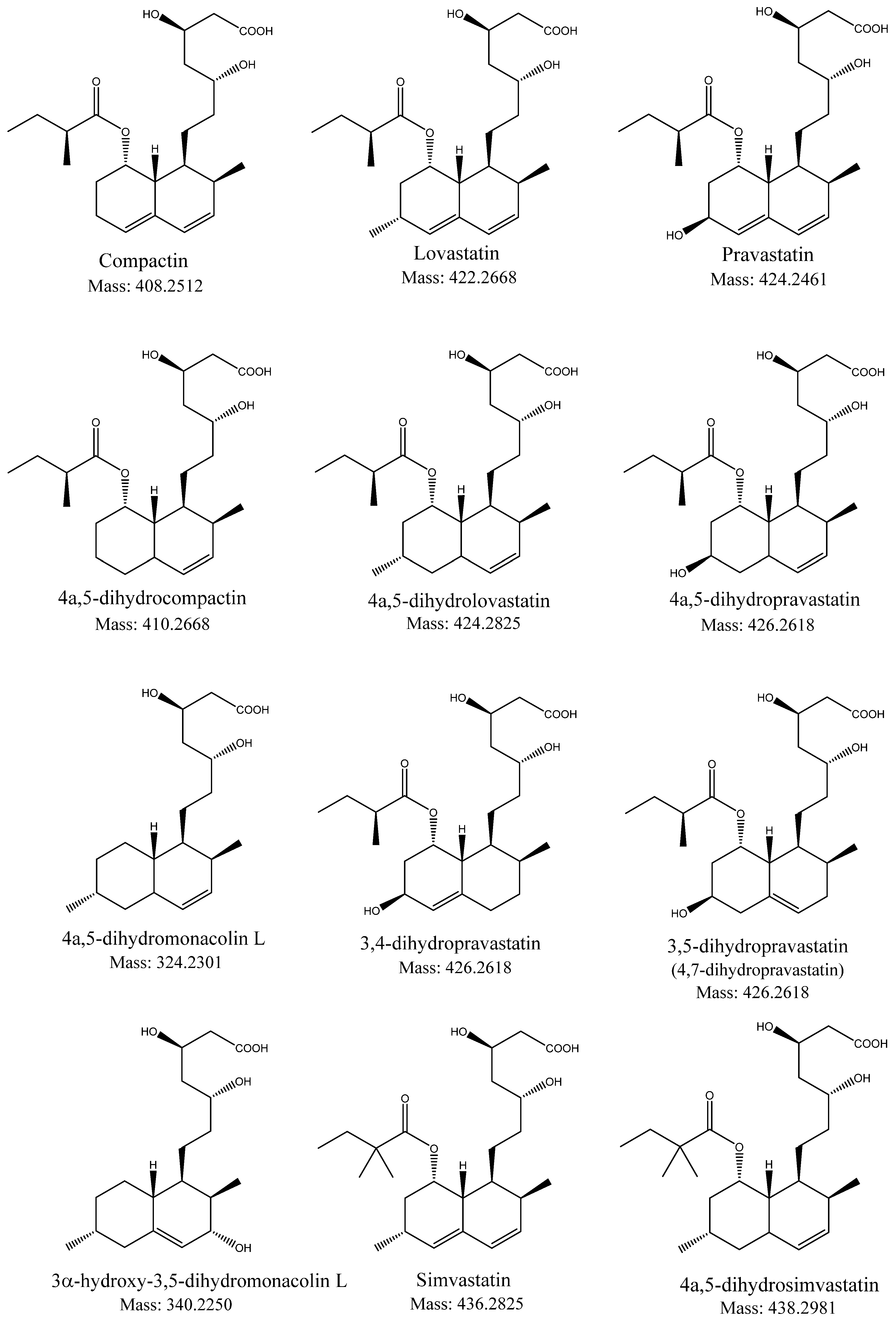
:1. Introduction
2. Results
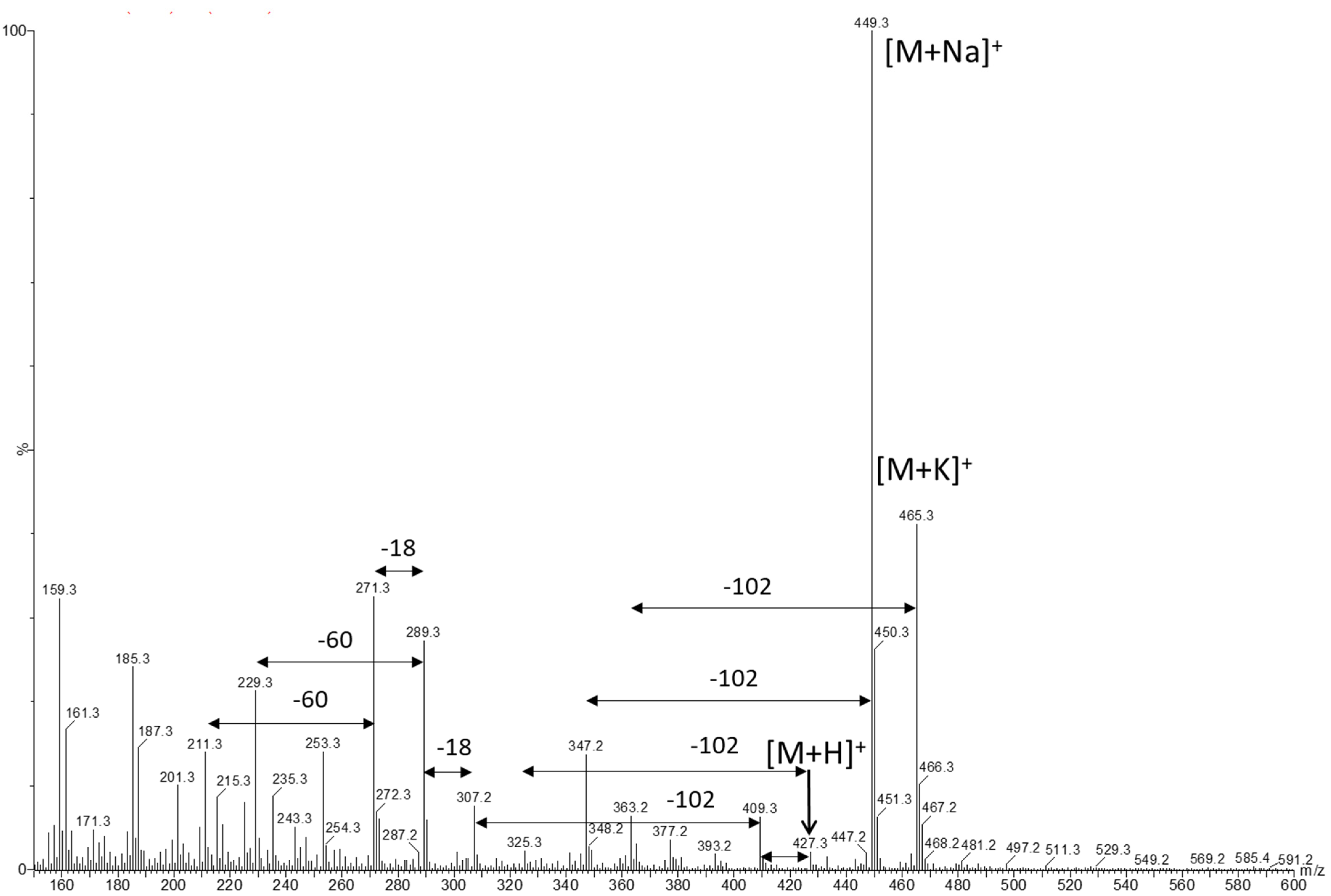
2.1. LC-MS Analysis of Dihydropravastatin during Pravastatin Fermentation
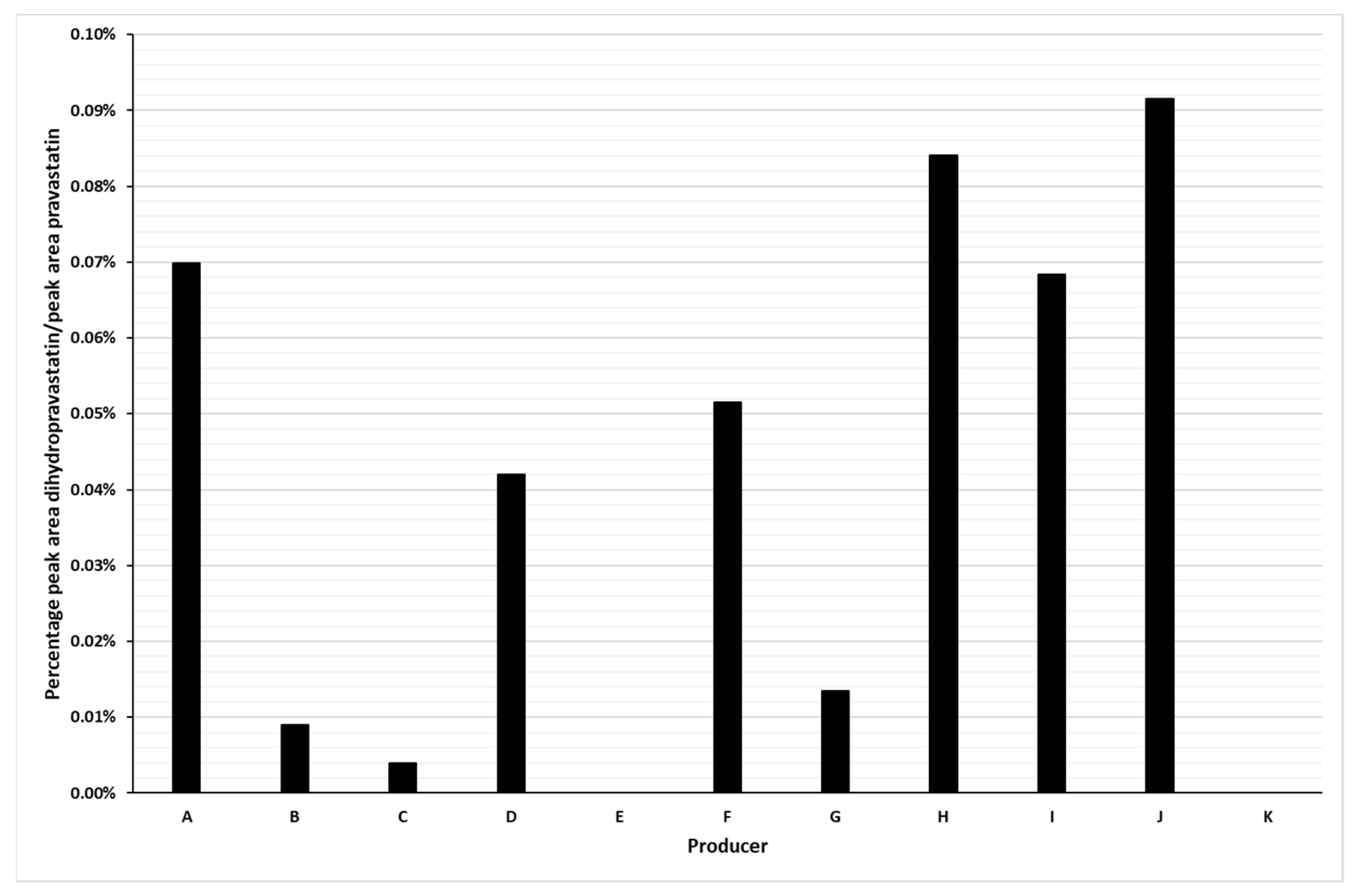
2.2. Detection of Dihydropravastatin Impurity in Pravastatin Formulations
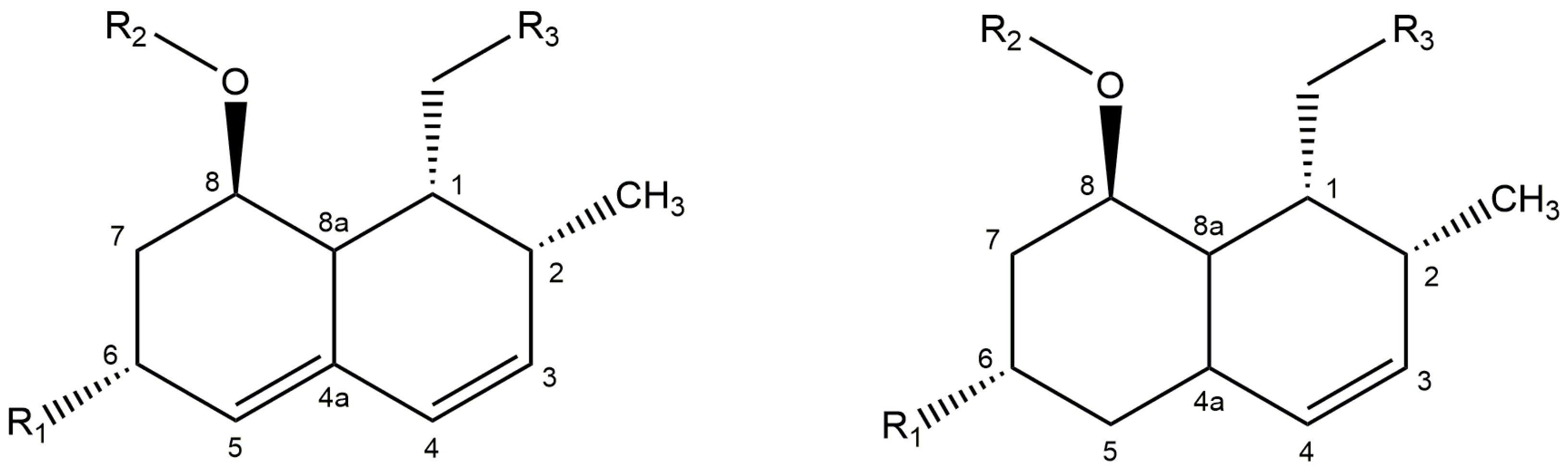
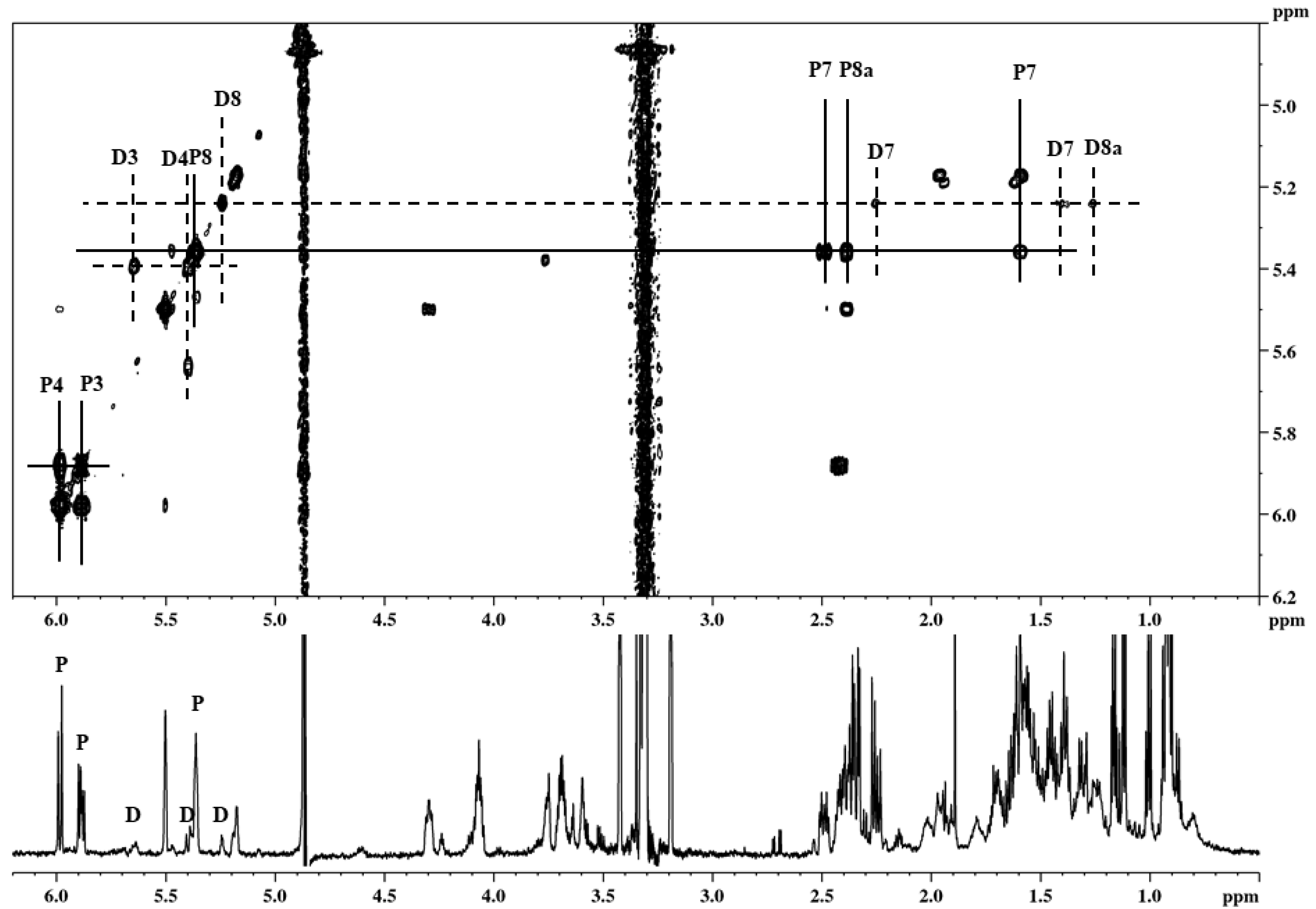
2.3. Identification of 4a,5-dihydropravastatin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | H3 | H4 | H8 | H8a | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| ppm | Δδ | ppm | Δδ | ppm | Δδ | ppm | Δδ | ||
| Monacolin L | 5.72 | 0.13 | 5.91 | 0.61 | (a) | (a) | (a) | (a) | [19] |
| 4a,5 dihydromonacolin L (Δ3,4) | 5.59 | 5.30 | (a) | (a) | |||||
| Compactin | 5.71 | 0.11 | 5.95 | 0.52 | 5.33 | 0.13 | - | - | [1,15] |
| 4a,5 dihydrocompactin (Δ3,4) | 5.60 | 5.43 | 5.20 | 1.22 | |||||
| Lovastatin | 5.78 | 0.13 | 6.00 | 0.62 | 5.40 | 0.19 | 2.26 | 1.07 | [6,9] |
| 4a,5 dihydrolovastatin (Δ3,4) | 5.65 | 5.38 | 5.21 | 1.19 | |||||
| Lovastatin | 5.78 | 0.14 | 5.99 | 0.60 | 5.38 | 0.20 | 2.26 | 1.07 | [7] |
| 4a,5 dihydrolovastatin (Δ3,4) | 5.64 | 5.39 | 5.18 | 1.19 | |||||
| Lovastatin | 5.84 | 0.15 | 6.01 | 0.59 | 5.35 | 0.20 | 2.37 | - | [20] (b) |
| 4a,5 dihydrolovastatin (Δ3,4) | 5.69 | 5.42 | 5.15 | - | |||||
| Lovastatin | - | - | - | - | - | - | - | - | [18] (d) |
| 3,5 dihydrolovastatin (Δ4,4a) | - | 5.48 | 5.28 | - | |||||
| Simvastatin | 5.78 | 0.13 | 5.99 | 0.60 | 5.37 | 0.18 | 2.26 | 1.06 | This study |
| 4a,5 dihydrosimvastatin (Δ3,4) | 5.65 | 5.39 | 5.19 | 1.20 | |||||
| Pravastatin | 5.88 | 3.80 | 5.99 | 0.45 | 5.40 | 0.04 | 2.32 | 0.50 | [11] |
| 3,5 Dihydro pravastatin (Δ4,4a) | 2.08 | 5.54 | 5.36 | 1.82 | |||||
| Pravastatin | - | - | - | - | - | - | - | - | [10] |
| 3,5 Dihydro pravastatin (Δ4,4a) | 2.06 | 5.51 | 5.33 | - | |||||
| Pravastatin | - | - | - | - | - | - | - | - | [17] (d) |
| 4a,5 dihydropravastatin lacton (Δ3,4) | 5.64 | 5.41 | 5.30 | - | |||||
| Pravastatin | 5.91 | 0.23 | 6.00 | 0.59 | 5.38 | 0.13 | 2.38 | 1.09 | This study (c) |
| 4a,5 dihydropravastatin (Δ3,4) | 5.68 | 5.41 | 5.25 | 1.27 | |||||
3. Discussion
3.1. Biosynthetic Pathway
3.2. Biological Activity of Dihydrostatins
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. LC-MS Analysis
4.3. NMR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brown, A.G.; Smale, T.C.; King, T.J.; Hasenkamp, R.; Thompson, R.H. Crystal and molecular structure of compactin, a new antifungal metabolite from Penicillium brevicompactum. J. Chem. Soc. Perkin Trans. I 1976, 11, 1165–1170. [Google Scholar] [CrossRef]
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B and ML-236C, new inhibitors of cholesterogenesis produced by Penicilium citrinum. J. Antibiot. 1976, 29, 1346–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, E.F.; Santos-Martins, D.; Ribeiro, A.M.; Brás, N.F.; Cerqueira, N.S.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. HMG-CoA Reductase inhibitors: An updated review of patents of novel compounds and formulations (2011–2015). Expert Opin. Ther. Pat. 2016, 26, 1257–1272. [Google Scholar] [CrossRef]
- Gullo, V.P.; Goegelman, R.T.; Putter, I.; Lam, Y.-K. High-performance liquid chromatographic analysis of derivatized hypocholesteremic agents from fermentation broths. J. Chromatogr. 1981, 212, 234–238. [Google Scholar] [CrossRef]
- Albers-Schönberg, G.; Joshua, H.; Lopez, M.B.; Hensens, O.D.; Springer, J.P.; Chen, J.; Ostrove, S.; Hoffman, C.H.; Alberts, A.W.; Patchett, A.A. Dihydromevinolin, a potent hypocholesterolemic metabolite produced by Aspergillus terreus. J. Antibiot. 1981, 34, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Lankhorst, P.P.; Poot, M.M.; de Lange, M.P.A. Quantitative determination of lovastatin and dihydrolovastatin by means of 1H-NMR spectroscopy. Pharmacop. Forum. 1996, 22, 2414–2422. [Google Scholar]
- Li, J.; Huang, H.-W.; Zhang, H.; Li, T.; Shi, Y.-Q. The impurity profiling of simvastatin and its tablets by UPLC-MS/MS. Yaoxue Xuebao 2014, 49, 672–678. [Google Scholar]
- Moore, R.N.; Bigam, G.; Chan, J.K.; Hogg, A.M.; Nakashima, T.T.; Vederas, J.C. Biosynthesis of the hypocholesterolemic agent mevinolin by Aspergillus terreus. Determination of the origin of carbon, hydrogen, and oxygen atoms by 13C NMR and mass spectrometry. J. Am. Chem. Soc. 1985, 107, 3694–3701. [Google Scholar] [CrossRef]
- Li, J.; Li, H.; Zhang, L. Isolation, structural identification and bioactivity of 4,7 dihydropravastatin. Chin. J. Antibiot. 2011, 7, 526–529. [Google Scholar]
- Bacher, M.; Baumann, K.; Knapp, H.; Steck, A.; Teibl, S. Complete assignment of 1H and 13C NMR data of pravastatin derivatives. Magn. Reson. Chem. 2009, 47, 71–83. [Google Scholar] [CrossRef]
- Barriuso, J.; Nguyen, D.T.; Li, J.W.-H.; Roberts, J.N.; MacNevin, G.; Chaytor, J.L.; Marcus, S.L.; Vederas, J.C.; Ro, D.-K. Double oxidation of the cyclic nonaketide dihydromonacolin L to monacolin J by a single cytochrome P450 monooxygenase, LovA. J. Am. Chem. Soc. 2011, 133, 8078–8081. [Google Scholar] [CrossRef]
- Wei, N.-Y.; Zhou, Y.; Yu, L.-J.; He, L.; Ning, B.-M. Analysis of related substances in pravastatin sodium and its preparations by UPLC-DAD-MS. Chin. J. Pharm. Anal. 2018, 38, 1539–1549. [Google Scholar]
- McLean, K.L.; Hans, M.; Meijrink, B.; van Scheppingen, W.B.; Vollebregt, A.; Tee, K.L.; van der Laan, J.-M.; Leys, D.; Munro, A.W.; van den Berg, M.A. Single-step fermentative production of the cholesterol-lowering drug pravastatin via reprogramming of Penicillium chrysogenum. Proc. Natl. Acad. Sci. USA 2015, 119, 2847–2852. [Google Scholar] [CrossRef] [Green Version]
- Lam, Y.K.T.; Gullo, V.P.; Goegelman, R.T.; Jorn, D.; Huang, L.; DeRiso, C.; Monaghan, R.L.; Putter, I. Dihydrocompactin, a new potent inhibitor of 3-hydroxy-3-methylglutaryl coenzyme-A reductase from Penicillium citrinum. J. Antibiot. 1981, 34, 614–616. [Google Scholar] [CrossRef] [Green Version]
- De Pater, R.M.; van Wijk, A.A.C.; Wnukowski, P. Diamine Salts of Carboxylic Acids. U.S. Patent Application WO2009121869, 8 October 2009. [Google Scholar]
- Bone, E.A.; Davidson, A.H.; Lewis, C.N.; Todd, R.S. Synthesis and biological evaluation of dihydroeptastatin, a novel inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Med. Chem. 1992, 35, 3388–3393. [Google Scholar] [CrossRef]
- DeCamp, A.E.; Verhoeven, T.R.; Shinkai, I. Synthesis of (+)-dihydromevinolin by selective reduction of mevinolin. J. Org. Chem. 1989, 54, 3207–3208. [Google Scholar] [CrossRef]
- Endo, A.; Hasumi, K.; Nakamura, T.; Kunishima, M.; Masuda, M. Dihydromonacolin L and monacolin X, new metabolites those inhibit cholesterol biosynthesis. J. Antibiot. 1985, 38, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Hachem, R.; Assemat, G.; Balayssac, S.; Martins-Froment, N.; Gilard, V.; Martino, P.; Malet-Martino, M. Comparative chemical profiling and monacolins quantification in red yeast rice dietary supplements by 1H-NMR and UHPLC-DAD-MS. Molecules 2020, 25, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, J.; Auclair, K.; Kendrew, S.G.; Park, C.; Vederas, J.C.; Hutchinson, C.R. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 1999, 284, 1368–1372. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, J.L.; Auclair, K.; Kennedy, J.; Hutchinson, C.R.; Vederas, J.C. Transformations of cyclic nonaketides by Aspergillus terreus mutants blocked for lovastatin biosynthesis at the lovA and lovC genes. Org. Biomol. Chem. 2003, 1, 50–59. [Google Scholar] [CrossRef]
- Abe, Y.; Suzuki, T.; Ono, C.; Iwamoto, K.; Hosobuchi, M.; Yoshikawa, H. Molecular cloning and characterization of an ML-236B (compactin) biosynthetic gene cluster in Penicillium citrinum. Mol. Genet. Genom. 2002, 267, 636–646. [Google Scholar] [CrossRef]
- Zong, H.; Zhuge, B.; Lu, X.; Huo, X.; Fang, H.; Song, J.; Sun, J. Characterization of a novel cytochrome P450 from Amycolatopsis Sp. CGMCC1149 for hydroxylation of lovastatin. Biotechnol. Appl. Biochem. 2015, 62, 9–16. [Google Scholar] [CrossRef]
- Phainuphong, P.; Rukachaisirikul, V.; Saithong, S.; Phongpaichit, S.; Bowornwiriyapan, K.; Muanprasat, C.; Srimaroeng, C.; Duangjai, A.; Sakayaroj, J. Lovastatin analogues from the soil-derived fungus Aspergillus sclerotiorum PSU-RSPG178. J. Nat. Prod. 2016, 79, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Heathcock, C.H.; Hadley, C.R.; Rosen, T.; Theisen, P.D.; Hecker, S.J. Total synthesis and biological evaluation of structural analogues of compactin and dihydromevinolin. J. Med. Chem. 1987, 30, 1858–1873. [Google Scholar] [CrossRef] [PubMed]
- Quion, J.A.; Jones, P.H. Clinical pharmacokinetics of pravastatin. Clin. Pharmacokinet. 1994, 27, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Joshua, H.; Schwartz, M.S.; Wilson, K.E. L-669,262, a potent HMG-CoA reductase inhibitor. J. Antibiot. 1991, 44, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Guan, S.; Zhang, L. Hypolipidemic and antioxidant activities of 4,5,6,7-tetrahydropravastatin sodium as a novel HMG-CoA reductase inhibitor in high-fat diet rats. Lat. Am. J. Pharm. 2016, 35, 1297–1303. [Google Scholar]
- Hirth, D. A New Combined LC (ESI+) MS/MS QTOF Impurity Fingerprinting and Chemometrics Approach for Discriminating Active Pharmaceutical Ingredient Origins: Example of Simvastatin. Analytical Chemistry Dumas-00960820. Available online: https://dumas.ccsd.cnrs.fr/dumas-00960820/document (accessed on 8 July 2011).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Scheppingen, W.B.; Lankhorst, P.P.; Hans, M.; van den Berg, M.A. Detection of 4a,5-dihydropravastatin as Impurity in the Cholesterol Lowering Drug Pravastatin. Molecules 2021, 26, 4685. https://doi.org/10.3390/molecules26154685
van Scheppingen WB, Lankhorst PP, Hans M, van den Berg MA. Detection of 4a,5-dihydropravastatin as Impurity in the Cholesterol Lowering Drug Pravastatin. Molecules. 2021; 26(15):4685. https://doi.org/10.3390/molecules26154685
Chicago/Turabian Stylevan Scheppingen, Wibo B., Peter P. Lankhorst, Marcus Hans, and Marco A. van den Berg. 2021. "Detection of 4a,5-dihydropravastatin as Impurity in the Cholesterol Lowering Drug Pravastatin" Molecules 26, no. 15: 4685. https://doi.org/10.3390/molecules26154685
APA Stylevan Scheppingen, W. B., Lankhorst, P. P., Hans, M., & van den Berg, M. A. (2021). Detection of 4a,5-dihydropravastatin as Impurity in the Cholesterol Lowering Drug Pravastatin. Molecules, 26(15), 4685. https://doi.org/10.3390/molecules26154685