What Is the Mechanism Driving the Reduction of Cardiovascular Events from Glucagon-like Peptide-1 Receptor Agonists?—A Mini Review



Abstract
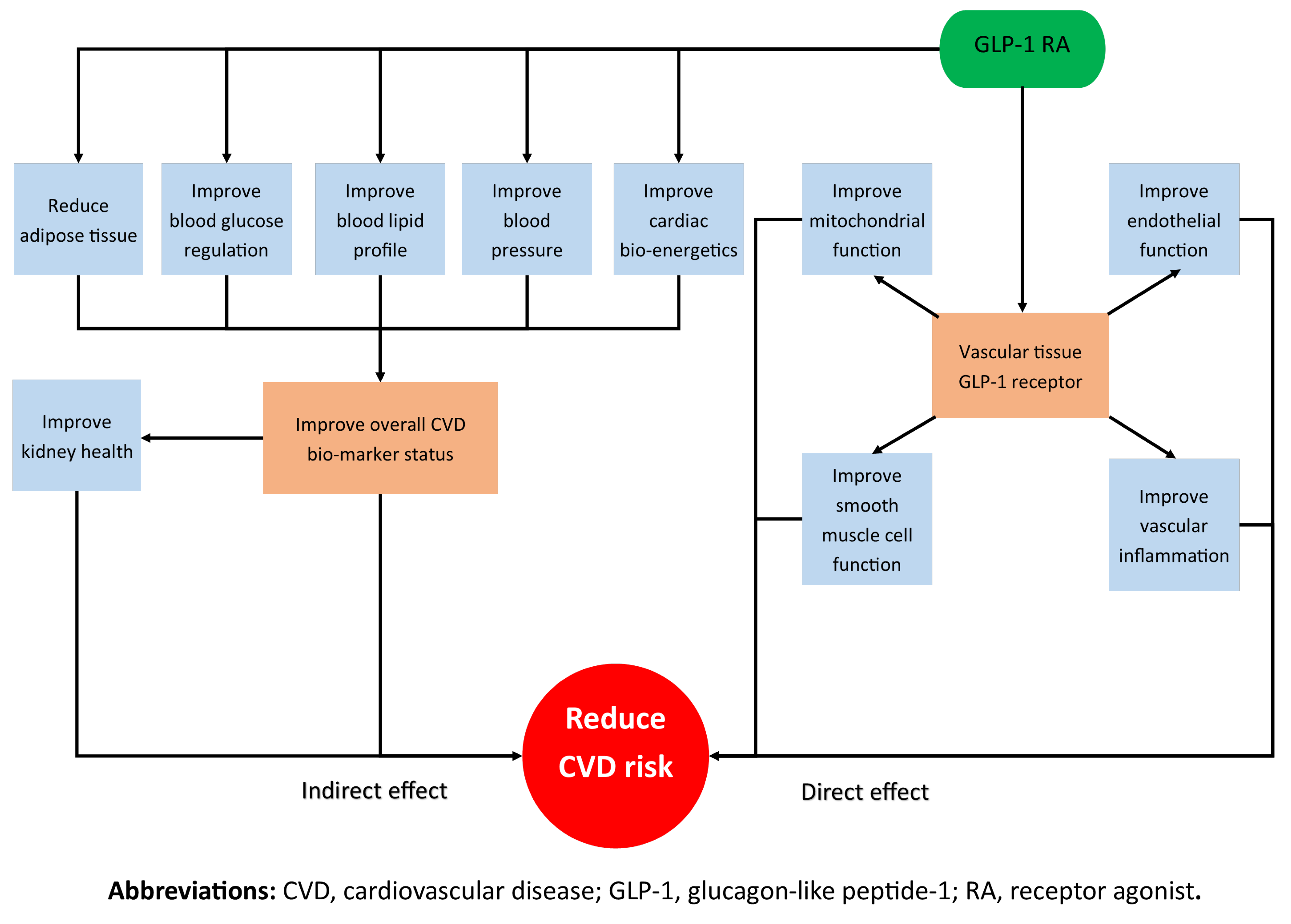
:1. Introduction
2. Indirect Effects of GLP-1 RAs on CVD Risk Reduction
2.1. Kidneys
2.2. Blood Glucose, BP, Lipids and Weight
2.3. Myocardium
3. Direct Effects of GLP-1 RAs on CVD Risk Reduction
3.1. Immune Response
3.2. Vascular Smooth Muscle Cells (VSMC)
3.3. Mitochondria
3.4. Endothelium
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | 5′-AMP-activated protein kinase |
| ANG II | Angiotensin II |
| APN | adipose adiponectin |
| BP | Blood pressure |
| CVOTs | Cardiovascular outcome trials |
| CV | Cardiovascular |
| CVD | Cardiovascular disease |
| DPP-4 | Dipeptidyl peptidase-4 |
| ERK | Extracellular signal-regulated kinase |
| ET-1 | Endothelin-1 |
| GLP-1 | Glucagon-like peptide-1 |
| GLP-1R | Glucagon-like peptide-1 receptor |
| KLF2 | Krüppel-like factor 2 |
| LOX-1 | Lectin-like oxidised low-density lipoprotein receptor-1 |
| MACE | Major adverse cardiovascular events |
| MI | Myocardial infarction |
| MMPs | Matrix metalloproteinases |
| NF | Nuclear factor |
| ox-LDL | Oxidised low-density lipoprotein |
| PDGF | Platelet-derived growth factor |
| RAs | Receptor agonists |
| ROS | Reactive oxygen species |
| T2D | Type 2 diabetes |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TNF | Tumour necrosis factor |
| VSMC | Vascular smooth muscle cells |
References
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2008, 26, 77–82. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of medical care in diabetes—2020. Diabetes Care 2020, 43, S98–S110. [Google Scholar] [CrossRef] [Green Version]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Furtado, R.H.M.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; et al. Comparison of the effects of glucagon-like peptide receptor agonists and sodium-glucose cotransporter 2 inhibitors for prevention of major adverse cardiovascular and renal outcomes in type 2 diabetes mellitus. Circulation 2019, 139, 2022–2031. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. 1), 5–21. [Google Scholar] [CrossRef]
- Sharma, D.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements & challenges. Biomed. Pharmacother. 2018, 108, 952–962. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Køber, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B., Sr.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Marso, S.P.; Christiansen, E.; Calanna, S.; Rasmussen, S.; Buse, J.B. Hypoglycemia, cardiovascular outcomes, and death: The LEADER experience. Diabetes Care 2018, 41, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.P.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and renal outcomes with efpeglenatide in type 2 diabetes. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine (U.S.). A Study of Taspoglutide in Patients with Inadequately Controlled Diabetes Mellitus Type 2 and Cardiovascular Disease. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01018173 (accessed on 2 August 2021).
- Seshasai, S.R.K.; Bennett, R.L.; Petrie, J.R.; Bengus, M.; Ekman, S.; Dixon, M.; Herz, M.; Buse, J.B.; Ray, K.K. Cardiovascular safety of the glucagon-like peptide-1 receptor agonist taspoglutide in people with type 2 diabetes: An individual participant data meta-analysis of randomized controlled trials. Diabetes. Obes. Metab. 2015, 17, 505–510. [Google Scholar] [CrossRef]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). A Study of Tirzepatide (LY3298176) Compared with Dulaglutide on Major Cardiovascular Events in Participants With Type 2 Diabetes. Available online: https://clinicaltrials.gov/ct2/show/NCT04255433 (accessed on 2 August 2021).
- Alonso, N.; Julián, M.T.; Puig-Domingo, M.; Vives-Pi, M. Incretin hormones as immunomodulators of atherosclerosis. Front. Endocrinol. 2012, 3, 112. [Google Scholar] [CrossRef] [Green Version]
- Matarese, A.; Gambardella, J.; Lombardi, A.; Wang, X.; Santulli, G. miR-7 regulates GLP-1-mediated insulin release by targeting β-arrestin 1. Cells 2020, 9, 1621. [Google Scholar] [CrossRef]
- Vedantham, S.; Kluever, A.K.; Deindl, E. Is there a chance to promote arteriogenesis by DPP4 inhibitors even in type 2 diabetes? A critical review. Cells 2018, 7, 181. [Google Scholar] [CrossRef] [Green Version]
- Cox, E.J.; Alicic, R.Z.; Neumiller, J.J.; Tuttle, K.R. Clinical evidence and proposed mechanisms for cardiovascular and kidney benefits from glucagon-like peptide-1 receptor agonists. US Endocrinol. 2020, 16, 80–87. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Pantalone, K.M.; Munir, K.; Hasenour, C.M.; Atisso, C.M.; Varnado, O.J.; Maldonado, J.M.; Konig, M. Cardiovascular outcomes trials with glucagon-like peptide-1 receptor agonists: A comparison of study designs, populations and results. Diabetes Obes. Metab. 2020, 22, 2209–2226. [Google Scholar] [CrossRef]
- van Baar, M.J.B.; van der Aart, A.B.; Hoogenberg, K.; Joles, J.A.; Heerspink, H.J.L.; van Raalte, D.H. The incretin pathway as a therapeutic target in diabetic kidney disease: A clinical focus on GLP-1 receptor agonists. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018819865398. [Google Scholar] [CrossRef]
- Skov, J. Effects of GLP-1 in the kidney. Rev. Endocr. Metab. Disord. 2014, 15, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Takashi, Y. GLP-1 Receptor Agonists in Diabetic Kidney Disease: From Clinical Outcomes to Mechanisms. Front. Pharmacol. 2020, 11, 967. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). A Research Study to See How Semaglutide Works Compared to Placebo in People with Type 2 Diabetes and Chronic Kidney Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03819153 (accessed on 2 August 2021).
- Colhoun, H.M.; Hasenour, C.; Riddle, M.C.; Branch, K.; Konig, M.; Atisso, C.; Lakshmanan, M.; Mody, R.; Gerstein, H.C. 924-p: Exploring potential mediators of the cardiovascular benefit of dulaglutide in REWIND. Diabetes 2020, 69, 924-P. [Google Scholar] [CrossRef]
- Turnbull, F.M.; Abraira, C.; Anderson, R.J.; Byington, R.P.; Chalmers, J.P.; Duckworth, W.C.; Evans, G.W.; Gerstein, H.C.; Holman, R.R.; Moritz, T.E.; et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia 2009, 52, 2288–2298. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.E.; O’Keefe, J.H.; Bell, D.S.H.; Schwartz, S.S. Insulin therapy increases cardiovascular risk in type 2 diabetes. Prog. Cardiovasc. Dis. 2017, 60, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Vella, A. Effects of GLP-1 on appetite and weight. Rev. Endocr. Metab. Disord. 2014, 15, 181–187. [Google Scholar] [CrossRef]
- Pratley, R.E.; Aroda, V.R.; Lingvay, I.; Lüdemann, J.; Andreassen, C.; Navarria, A.; Viljoen, A. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): A randomised, open-label, phase 3b trial. Lancet. Diabetes Endocrinol. 2018, 6, 275–286. [Google Scholar] [CrossRef]
- Brown, J.D.; Buscemi, J.; Milsom, V.; Malcolm, R.; O’Neil, P.M. Effects on cardiovascular risk factors of weight losses limited to 5–10. Transl. Behav. Med. 2016, 6, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimihodimos, V.; Elisaf, M. Incretins and lipid metabolism. Curr. Med. Chem. 2018, 25, 2133–2139. [Google Scholar] [CrossRef]
- Liu, L.; Liu, J.; Huang, Y. Protective Effects of Glucagon-like Peptide 1 on Endothelial Function in Hypertension. J. Cardiovasc. Pharmacol. 2015, 65, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Al Batran, R.; Almutairi, M.; Ussher, J.R. Glucagon-like peptide-1 receptor mediated control of cardiac energy metabolism. Peptides 2018, 100, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Drucker, D.J. Cardiovascular actions of incretin-based therapies. Circ. Res. 2014, 114, 1788–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV protection in the EMPA-REG OUTCOME trial: A “thrifty substrate” hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Kato, E.T.; Das, S.R.; McGuire, D.K. Antihyperglycemic therapies and cardiovascular outcomes in patients with type 2 diabetes mellitus: State of the art and future directions. Trends Cardiovasc. Med. 2020, 31, 101–108. [Google Scholar] [CrossRef]
- Sudo, M.; Li, Y.; Hiro, T.; Takayama, T.; Mitsumata, M.; Shiomi, M.; Sugitani, M.; Matsumoto, T.; Hao, H.; Hirayama, A. Inhibition of plaque progression and promotion of plaque stability by glucagon-like peptide-1 receptor agonist: Serial in vivo findings from iMap-IVUS in Watanabe heritable hyperlipidemic rabbits. Atherosclerosis 2017, 265, 283–291. [Google Scholar] [CrossRef]
- Koshibu, M.; Mori, Y.; Saito, T.; Kushima, H.; Hiromura, M.; Terasaki, M.; Takada, M.; Fukui, T.; Hirano, T. Antiatherogenic effects of liraglutide in hyperglycemic apolipoprotein E-null mice via AMP-activated protein kinase-independent mechanisms. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E895–E907. [Google Scholar] [CrossRef]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakipovski, G.; Rolin, B.; Nøhr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sørensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE(−/−) and LDLr(−/−) mice by a mechanism that includes inflammatory pathways. JACC Basic Transl. Sci. 2018, 3, 844–857. [Google Scholar] [CrossRef]
- Gaspari, T.; Welungoda, I.; Widdop, R.E.; Simpson, R.W.; Dear, A.E. The GLP-1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an ApoE(−/−) mouse model. Diabetes Vasc. Dis. Res. 2013, 10, 353–360. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Matsuo, Y.; Yamakage, H.; Masuda, S.; Terada, Y.; Muranaka, K.; Wada, H.; Hasegawa, K.; Shimatsu, A.; Satoh-Asahara, N. Differential effects of GLP-1 receptor agonist on foam cell formation in monocytes between non-obese and obese subjects. Metabolism 2016, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, D.; Fujiwara, Y.; Komohara, Y.; Mizuta, H.; Takeya, M. Glucagon-like peptide-1 (GLP-1) induces M2 polarization of human macrophages via STAT3 activation. Biochem. Biophys. Res. Commun. 2012, 425, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Bruen, R.; Curley, S.; Kajani, S.; Crean, D.; O’Reilly, M.E.; Lucitt, M.B.; Godson, C.G.; McGillicuddy, F.C.; Belton, O. Liraglutide dictates macrophage phenotype in apolipoprotein E null mice during early atherosclerosis. Cardiovasc. Diabetol. 2017, 16, 143. [Google Scholar] [CrossRef] [Green Version]
- Bruen, R.; Curley, S.; Kajani, S.; Lynch, G.; O’Reilly, M.E.; Dillon, E.T.; Brennan, E.P.; Barry, M.; Sheehan, S.; McGillicuddy, F.C.; et al. Liraglutide attenuates preestablished atherosclerosis in apolipoprotein e-deficient mice via regulation of immune cell phenotypes and proinflammatory mediators. J. Pharmacol. Exp. Ther. 2019, 370, 447–458. [Google Scholar] [CrossRef]
- Bułdak, Ł.; Łabuzek, K.; Bułdak, R.J.; Machnik, G.; Bołdys, A.; Okopień, B. Exenatide (a GLP-1 agonist) improves the antioxidative potential of in vitro cultured human monocytes/macrophages. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.N.; Xie, B.D.; Sun, L.; Chen, W.; Jiang, S.L.; Liu, W.; Bian, F.; Tian, H.; Li, R.K. Phenotypic switching of vascular smooth muscle cells in the ’normal region’ of aorta from atherosclerosis patients is regulated by miR-145. J. Cell. Mol. Med. 2016, 20, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, M.; Zhou, T.; Shen, Q.; Qin, X. Exendin-4 promotes the vascular smooth muscle cell re-differentiation through AMPK/SIRT1/FOXO3a signaling pathways. Atherosclerosis 2018, 276, 58–66. [Google Scholar] [CrossRef]
- Jojima, T.; Uchida, K.; Akimoto, K.; Tomotsune, T.; Yanagi, K.; Iijima, T.; Suzuki, K.; Kasai, K.; Aso, Y. Liraglutide, a GLP-1 receptor agonist, inhibits vascular smooth muscle cell proliferation by enhancing AMP-activated protein kinase and cell cycle regulation, and delays atherosclerosis in ApoE deficient mice. Atherosclerosis 2017, 261, 44–51. [Google Scholar] [CrossRef]
- Nagayama, K.; Kyotani, Y.; Zhao, J.; Ito, S.; Ozawa, K.; Bolstad, F.A.; Yoshizumi, M. Exendin-4 prevents vascular smooth muscle cell proliferation and migration by angiotensin ii via the inhibition of ERK1/2 and JNK signaling pathways. PLoS ONE 2015, 10, e0137960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Ji, Y.; Jiang, X.; Zhou, L.; Xu, Y.; Li, Y.; Jiang, W.; Meng, P.; Liu, X. Liraglutide attenuates high glucose-induced abnormal cell migration, proliferation, and apoptosis of vascular smooth muscle cells by activating the GLP-1 receptor, and inhibiting ERK1/2 and PI3K/Akt signaling pathways. Cardiovasc. Diabetol. 2015, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.; Pushpakumar, S.; Muradashvili, N.; Kundu, S.; Tyagi, S.C.; Sen, U. Regulation and involvement of matrix metalloproteinases in vascular diseases. Front. Biosci. 2016, 21, 89–118. [Google Scholar] [CrossRef] [Green Version]
- Garczorz, W.; Gallego-Colon, E.; Kosowska, A.; Kłych-Ratuszny, A.; Woźniak, M.; Marcol, W.; Niesner, K.J.; Francuz, T. Exenatide exhibits anti-inflammatory properties and modulates endothelial response to tumor necrosis factor α-mediated activation. Cardiovasc. Ther. 2018, 36, e12317. [Google Scholar] [CrossRef]
- Gallego-Colon, E.; Klych-Ratuszny, A.; Kosowska, A.; Garczorz, W.; Aghdam, M.R.F.; Wozniak, M.; Francuz, T. Exenatide modulates metalloproteinase expression in human cardiac smooth muscle cells via the inhibition of Akt signaling pathway. Pharmacol. Rep. 2018, 70, 178–183. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Q.; Pu, H.; Wei, Q.; Duan, M.; Zhang, C.; Jiang, T.; Shou, X.; Zhang, J.; Yang, Y. Adiponectin improves NF-κB-mediated inflammation and abates atherosclerosis progression in apolipoprotein E-deficient mice. Lipids Health Dis. 2016, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Lei, Y.; Inoue, A.; Piao, L.; Hu, L.; Jiang, H.; Sasaki, T.; Wu, H.; Xu, W.; Yu, C.; et al. Exenatide mitigated diet-induced vascular aging and atherosclerotic plaque growth in ApoE-deficient mice under chronic stress. Atherosclerosis 2017, 264, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Wang, W.T.; Lee, S.S.; Kuo, Y.R.; Wang, Y.C.; Yen, S.J.; Lee, M.Y.; Yeh, J.L. Glucagon-like peptide-1 receptor agonist attenuates autophagy to ameliorate pulmonary arterial hypertension through Drp1/NOX- and Atg-5/Atg-7/Beclin-1/Lc3β pathways. Int. J. Mol. Sci. 2019, 20, 3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, G.; Zhang, D.; Liu, J.; Zhang, P.; Ye, L.; Lu, K.; Duan, Q.; Zheng, A.; Qin, S. Exenatide protects against hypoxia/reoxygenation-induced apoptosis by improving mitochondrial function in H9c2 cells. Exp. Biol. Med. 2014, 239, 414–422. [Google Scholar] [CrossRef]
- DeNicola, M.; Du, J.; Wang, Z.; Yano, N.; Zhang, L.; Wang, Y.; Qin, G.; Zhuang, S.; Zhao, T.C. Stimulation of glucagon-like peptide-1 receptor through exendin-4 preserves myocardial performance and prevents cardiac remodeling in infarcted myocardium. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E630–E643. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.Y.; Chen, Z.W.; Gao, Y.H.; Wang, X.X.; Ma, J.Y.; Chang, S.F.; Qian, J.Y.; Ge, J.B. Exenatide reduces tumor necrosis factor-α-induced apoptosis in cardiomyocytes by alleviating mitochondrial dysfunction. Chin. Med. J. 2015, 128, 3211–3218. [Google Scholar] [CrossRef]
- Dai, Y.; Mercanti, F.; Dai, D.; Wang, X.; Ding, Z.; Pothineni, N.V.; Mehta, J.L. LOX-1, a bridge between GLP-1R and mitochondrial ROS generation in human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2013, 437, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Zhu, F.; Zheng, H.; Zhou, Z.; Miao, P.; Zhao, L.; Mao, Z. Glucagon-like peptide-1 receptor agonist dulaglutide prevents ox-LDL-induced adhesion of monocytes to human endothelial cells: An implication in the treatment of atherosclerosis. Mol. Immunol. 2019, 116, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Li, Y.; Ou, D.; Yang, Q. The GLP-1 receptor agonist liraglutide protects against oxidized LDL-induced endothelial inflammation and dysfunction via KLF2. IUBMB Life 2019, 71, 1347–1354. [Google Scholar] [CrossRef]
- Gaspari, T.; Liu, H.; Welungoda, I.; Hu, Y.; Widdop, R.E.; Knudsen, L.B.; Simpson, R.W.; Dear, A.E. A GLP-1 receptor agonist liraglutide inhibits endothelial cell dysfunction and vascular adhesion molecule expression in an ApoE−/− mouse model. Diabetes Vasc. Dis. Res. 2011, 8, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Mehta, J.L.; Chen, M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa b activation. Cardiovasc. Drugs Ther. 2013, 27, 371–380. [Google Scholar] [CrossRef]


{kind=link}
| Author (Year) | Molecule | N | Hazard Ratio (95% CI) | p-Value |
|---|---|---|---|---|
| Pfeffer et al. (2015) [8] | Lixisenatide | 6068 | 1.02 (0.89–1.17) | 0.81 |
| Marso et al. (2016) [10] | Semaglutide | 3297 | 0.74 (0.58–0.95) | 0.02 * |
| Holman et al. (2017) [9] | Exenatide | 14,752 | 0.91 (0.83–1.00) | 0.06 |
| Hernandez et al. (2018) [11] | Albiglutide | 9463 | 0.78 (0.68–0.90) | 0.0006 * |
| Zinman et al. (2018) [12] | Liraglutide | 9340 | 0.87 (0.78–0.97) | 0.01 * |
| Gerstein et al. (2019) [7] | Dulaglutide | 9901 | 0.88 (0.79–0.99) | 0.026 * |
| Gerstein et al. (2021) [13] | Efpeglenatide | 4076 | 0.73 (0.58–0.92) | 0.007 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berndt, J.; Ooi, S.L.; Pak, S.C. What Is the Mechanism Driving the Reduction of Cardiovascular Events from Glucagon-like Peptide-1 Receptor Agonists?—A Mini Review. Molecules 2021, 26, 4822. https://doi.org/10.3390/molecules26164822
Berndt J, Ooi SL, Pak SC. What Is the Mechanism Driving the Reduction of Cardiovascular Events from Glucagon-like Peptide-1 Receptor Agonists?—A Mini Review. Molecules. 2021; 26(16):4822. https://doi.org/10.3390/molecules26164822
Chicago/Turabian StyleBerndt, Jared, Soo Liang Ooi, and Sok Cheon Pak. 2021. "What Is the Mechanism Driving the Reduction of Cardiovascular Events from Glucagon-like Peptide-1 Receptor Agonists?—A Mini Review" Molecules 26, no. 16: 4822. https://doi.org/10.3390/molecules26164822
APA StyleBerndt, J., Ooi, S. L., & Pak, S. C. (2021). What Is the Mechanism Driving the Reduction of Cardiovascular Events from Glucagon-like Peptide-1 Receptor Agonists?—A Mini Review. Molecules, 26(16), 4822. https://doi.org/10.3390/molecules26164822