
Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs




Abstract
:1. Introduction
2. Tumor Microenvironment and Signaling Pathways—Possible Targets of Oleanolic acid Derivatives
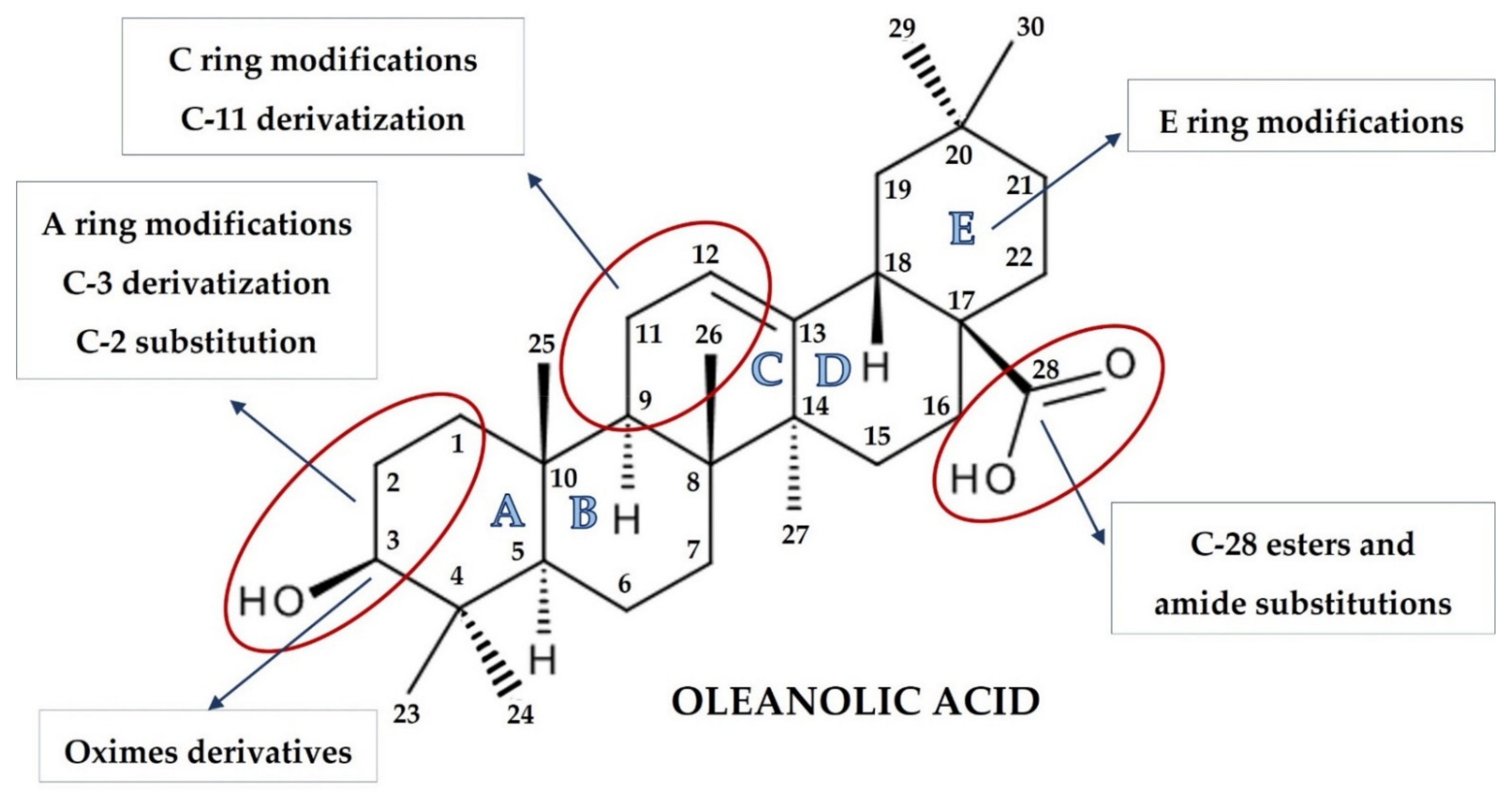
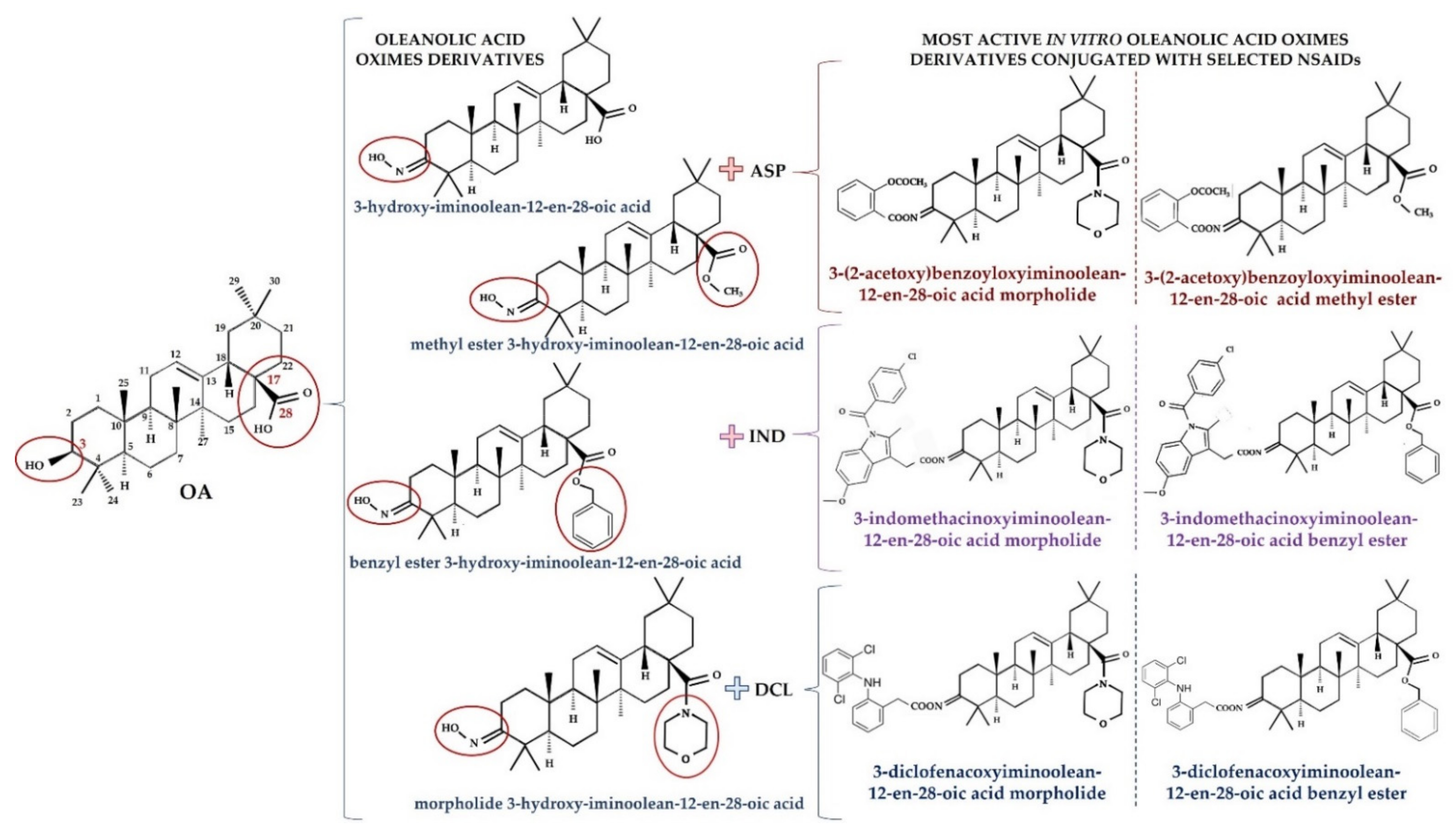
3. Overview of Oleanolic Acid Derivatives—Chemical Approach
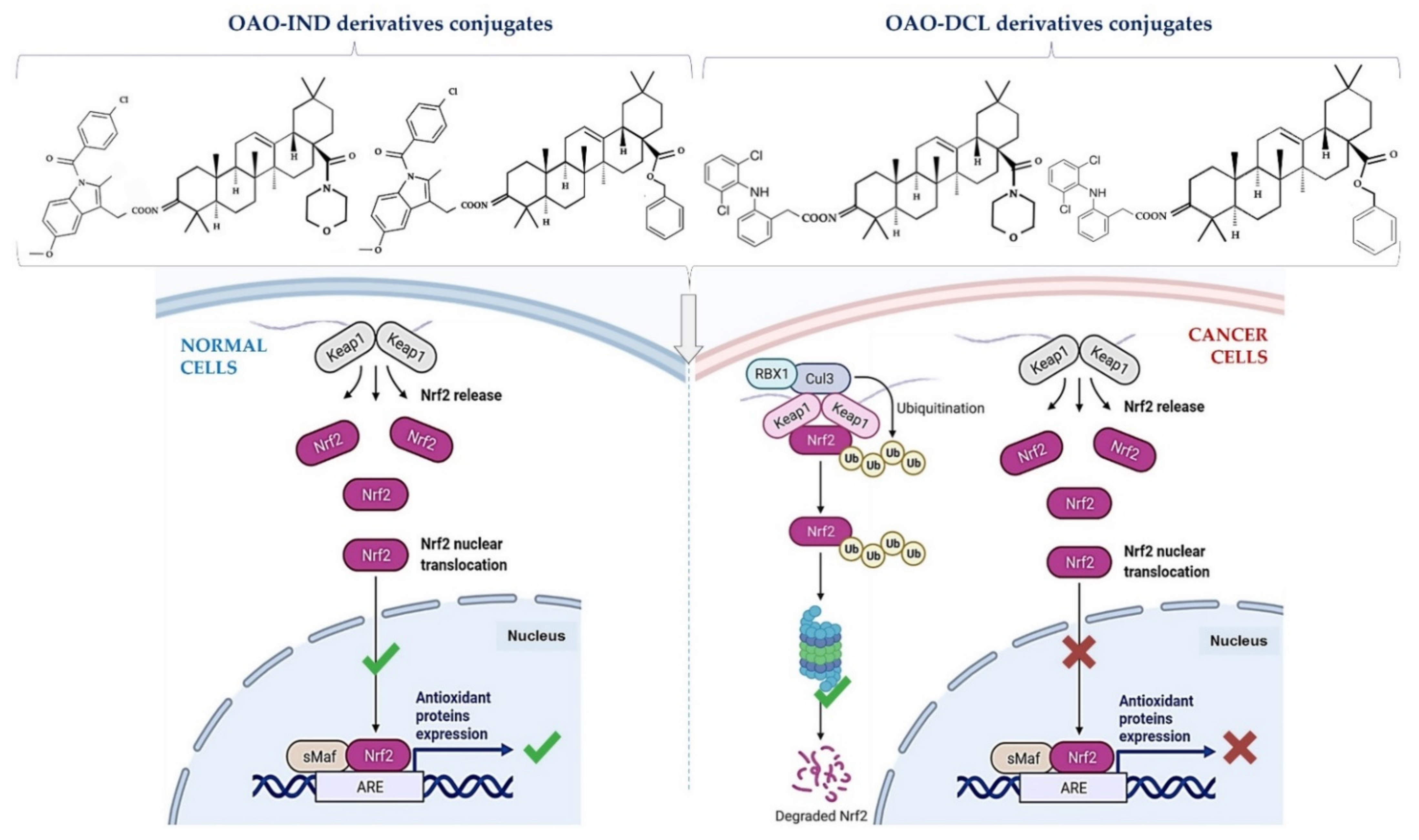
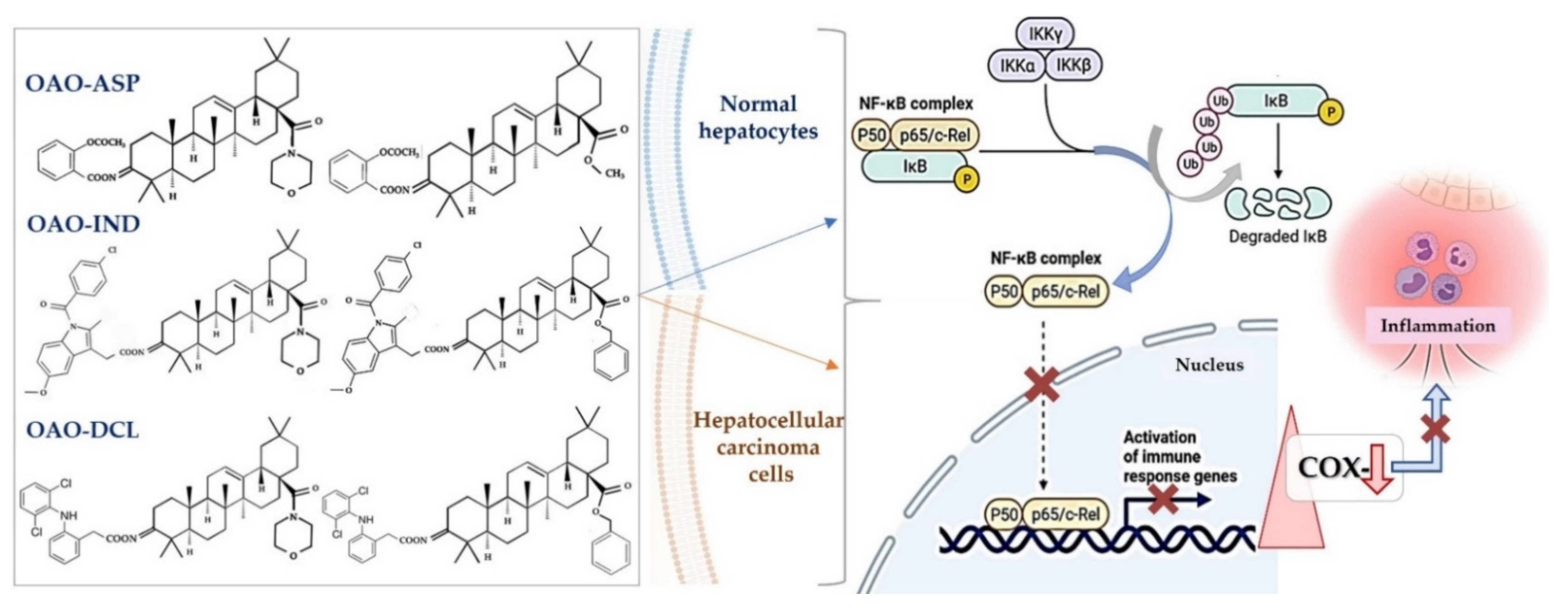
4. Docking Studies-Targeting Nrf2 and NF-κB
5. Biological Activity of Selected Synthetic OA Derivatives Which May Affect Cancer Development
6. Conjugation of Synthetic OA Derivatives May Enhance Their Anti-Cancer Potential
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Guinda, Á.; Rada, M.; Delgado, T.; Gutiérrez-Adánez, P.; Castellano, J.M. Pentacyclic triterpenoids from olive fruit and leaf. J. Agric. Food Chem. 2010, 58, 9685–9691. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants-rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [Green Version]
- Pollier, J.; Goossens, A. Oleanolic acid. Phytochemistry 2012, 77, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Pawełczyk, A.; Olender, D.; Sowa-Kasprzak, K.; Zaprutko, L. Hybrid compounds strategy in the synthesis of oleanolic acid skeleton-NSAID derivatives. Molecules 2016, 21, 420. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Liby, K.T.; Sporn, M.B. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef] [Green Version]
- Albini, A.; Tosetti, F.; Li, V.W.; Noonan, D.M.; Li, W.W. Cancer prevention by targeting angiogenesis. Nat. Rev. Clin. Oncol. 2012, 9, 498–509. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-Diclofenac as an anti-cancer agent. eCancerMedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Sig. Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Sig. Transduct. Target Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 2009, 4, a000067. [Google Scholar] [CrossRef] [Green Version]
- Rius-Pérez, S.; Pérez, S.; Martí-Andrés, P.; Monsalve, M.; Sastre, J. Nuclear Factor Kappa B Signaling Complexes in Acute Inflammation. Antioxid. Redox Signal. 2020, 3, 145–165. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently Activated Stat3 Maintains Constitutive NF-κB Activity in Tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.H.; Zhu, B.M.; Riedlinger, G.; Kang, K.; Hennighausen, L. The liver-specific tumor suppressor STAT5 controls expression of the reactive oxygen species-generating enzyme NOX4 and the proapoptotic proteins PUMA and BIM in mice. Hepatology 2012, 56, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in hepatocellular carcinoma: Role in cancer progression and chemoresistance. Cancers 2018, 12, 481. [Google Scholar] [CrossRef] [Green Version]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 3, 393–402. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 9, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Pantziarka, P.; Ghibelli, L.; Reichle, A. A computational model of tumor growth and anakoinosis. Front. Pharmacol. 2019, 10, 287. [Google Scholar] [CrossRef]
- Song, X.; Liu, C.C.; Hong, Y.R.; Zhu, X.C. Anti-cancer activity of novel oleanolic acid methyl ester derivative in HeLa cervical cancer cells is mediated through apoptosis induction and reactive oxygen species production. Bangladesh J. Pharmacol. 2015, 10, 896–902. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhou, M.; Duan, L.; Wang, W.; Zhang, J.; Wang, D.; Liang, X. Efficient synthesis and anti-fungal activity of oleanolic acid oxime esters. Molecules 2013, 18, 3615–3629. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk-Cwynar, B.; Zaprutko, L.; Marciniak, J.; Lewandowski, G.; Szulc, M.; Kaminska, E.; Wachowiak, N.; Mikolajczak, P.L. The analgesic and anti-inflammatory effect of new oleanolic acid acyloxyimino derivative. Eur. J. Pharm. Sci. 2012, 47, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Masullo, M.; Pizza, C.; Piacente, S. Oleanane derivatives for pharmaceutical use: A patent review (2000-2016). Expert Opin. Ther. Pat. 2016, 3, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Maitra, A.; Hruban, R.H.; Sporn, M.B. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev. Res. 2010, 3, 1427–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liby, K.T.; Yore, M.M.; Sporn, M.B. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer 2007, 5, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Yore, M.M.; Honda, T.; Gribble, G.W.; Dmitrovsky, E.; Sporn, T.A.; Sporn, M.B. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007, 67, 2414–2419. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Liu, J.; Wen, X.; Sun, H. Synthesis and cytotoxicity evaluation of oleanolic acid derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 2074–2077. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Paluszczak, J.; Szaefer, H.; Narożna, M.; Zaprutko, L.; Baer-Dubowska, W. Oleanolic acid oxime derivatives and their conjugates with aspirin modulate the NF-κB-mediated transcription in HepG2 hepatoma cells. Bioorg. Chem. 2019, 93, 103326. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins 2007, 67, 1010–1025. [Google Scholar] [CrossRef]
- Meng, X.; Waddington, J.C.; Tailor, A.; Lister, A.; Hamlett, J.; Berry, N.; Park, B.K.; Sporn, M.B. CDDO-imidazolide Targets Multiple Amino Acid Residues on the Nrf2 Adaptor, Keap1. J. Med. Chem. 2020, 63, 9965–9976. [Google Scholar] [CrossRef]
- Kamble, S.M.; Patel, H.M.; Goyal, S.N.; Noolvi, M.N.; Mahajan, U.B.; Ojha, S.; Patil, C.R. In silico Evidence for Binding of Pentacyclic Triterpenoids to Keap1-Nrf2 Protein-Protein Binding Site. Comb. Chem. Highthroughput Screen. 2017, 20, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Kucińska, M.; Kleszcz, R.; Kujawski, J.; Piotrowska-Kempisty, H.; Plewiński, A.; Murias, M.; Baer-Dubowska, W. Conjugation of Diclofenac with Novel Oleanolic Acid Derivatives Modulate Nrf2 and NF-κB Activity in Hepatic Cancer Cells and Normal Hepatocytes Leading to Enhancement of Its Therapeutic and Chemopreventive Potential. Pharmaceuticals 2021, 14, 688. [Google Scholar] [CrossRef]
- Ortega-Muñoz, M.; Rodríguez-Serrano, F.; De Los Reyes-Berbel, E.; Mut-Salud, N.; Hernández-Mateo, F.; Rodríguez-López, A.; Garrido, J.M.; López-Jaramillo, F.J.; Santoyo-González, F. Biological Evaluation and Docking Studies of Synthetic Oleanane-type Triterpenoids. ACS Omega 2018, 3, 11455–11468. [Google Scholar] [CrossRef]
- Patil, K.R.; Mohapatra, P.; Patel, H.M.; Goyal, S.N.; Ojha, S.; Kundu, C.N.; Patil, C.R. Pentacyclic Triterpenoids Inhibit IKKβ Mediated Activation of NF-κB Pathway: In Silico and In Vitro Evidences. PLoS ONE 2015, 10, e0125709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yore, M.M.; Liby, K.T.; Honda, T.; Gribble, G.W.; Sporn, M.B. The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol. Cancer Ther. 2006, 5, 3232–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Snowden, R.T.; Dyer, M.J.S.; Cohen, G.M. CDDO induces apoptosis via the intrinsic pathway in lymphoid cells. Leukemia 2004, 18, 948–952. [Google Scholar] [CrossRef] [Green Version]
- Koschmieder, S.; D’Alò, F.; Radomska, H.; Schöneich, C.; Ji, S.C.; Konopleva, M.; Kobayashi, S.; Levantini, E.; Suh, N.; Di Ruscio, A.; et al. CDDO induces granulocytic differentiation of myeloid leukemic blasts through translational up-regulation of p42 CCAAT enhancer-binding protein alpha. Blood 2007, 110, 3695–3705. [Google Scholar] [CrossRef] [Green Version]
- Suh, N.; Wang, Y.; Honda, T.; Gribble, G.W.; Dmitrovsky, E.; Hickey, W.F.; Maue, R.A.; Place, A.E.; Porter, D.M.; Spinella, M.J.; et al. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999, 59, 336–341. [Google Scholar]
- Shishodia, S.; Sethi, G.; Konopleva, M.; Andreeff, M.; Aggarwal, B.B. A Synthetic Triterpenoid, CDDO-Me, Inhibits IKBA Kinase and Enhances Apoptosis Induced by TNF and Chemotherapeutic Agents through Down-Regulation of Expression of Nuclear Factor KB Regulated Gene Products in Human Leukemic Cells. Clin. Cancer Res. 2006, 12, 1828–1838. [Google Scholar] [CrossRef] [Green Version]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef] [Green Version]
- Pitha-Rowe, I.; Liby, K.; Royce, D.; Sporn, M. Synthetic triterpenoids attenuate cytotoxic retinal injury: Cross-talk between Nrf2 and PI3K/AKT signaling through inhibition of the lipid phosphatase PTEN. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5339–5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Zhe, H.; Zhao, R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol. Cancer 2014, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- To, C.; Roy, A.; Chan, E.; Prado, M.A.M.; Di Guglielmo, G.M. Synthetic triterpenoids inhibit GSK3β activity and localization and affect focal adhesions and cell migration. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 1274–1284. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Dulchavsky, S.A.; Gautam, S.C. CDDO-Me inhibits proliferation, induces apoptosis, down-regulates Akt, mTOR, NF-kappaB and NF-kappaB-regulated anti-apoptotic and proangiogenic proteins in TRAMP prostate cancer cells. J. Exp. Ther. Oncol. 2008, 7, 31–39. [Google Scholar] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [CrossRef] [Green Version]
- C17-Heteroaryl Derivatives of Oleanolic Acid and Methods of Use. 2013. Available online: https://patents.google.com/patent/MX2015003078A/en (accessed on 19 May 2021).
- Narożna, M.; Krajka-Kuźniak, V.; Kleszcz, R.; Bednarczyk-Cwynar, B.; Szaefer, H.; Baer-Dubowska, W. Activation of the Nrf2 response by oleanolic acid oxime morpholide (3-hydroxyiminoolean-12-en-28-oic acid morpholide) is associated with its ability to induce apoptosis and inhibit proliferation in HepG2 hepatoma cells. Eur. J. Pharmacol. 2020, 883, 173307. [Google Scholar] [CrossRef]
- CDDO-Me Amino Acid Conjugates and Methods of Use. 2014. Available online: https://patents.google.com/patent/US9290455B2/en (accessed on 19 May 2021).
- Macasoi, I.; Pavel, I.Z.; Moaca, A.E.; Avram, S.; David, V.L.; Coricovac, D.; Mioc, A.; Spandidos, D.A.; Tsatsakis, A.; Soica, C.; et al. Mechanistic investigations of antitumor activity of a Rhodamine B-oleanolic acid derivative bioconjugate. Oncol. Rep. 2020, 44, 1169–1183. [Google Scholar] [CrossRef]
- Mo, W.B.; Su, C.H.; Huang, J.Y.; Liu, J.; Chen, Z.F.; Cheng, K.G. Synthesis of acyl oleanolic acid-uracil conjugates and their anti-tumor activity. Chem. Cent. J. 2016, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.S.Y. Role of Non-steroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharmacol. Sci. 2019, 2019, 3418975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S.S.; Girgis, A.S.; Honkanadavar, H.H.; George, R.F.; Srour, A.M. Synthesis of new ibuprofen hybrid conjugates as potential anti-inflammatory and analgesic agents. Future Med. Chem. 2020, 12, 1369–1386. [Google Scholar] [CrossRef]
- Bednarczyk-Cwynar, B.; Wachowiak, N.; Szulc, M.; Kamińska, E.; Bogacz, A.; Bartkowiak-Wieczorek, J.; Zaprutko, L.; Mikolajczak, P.L. Strong and Long-Lasting Antinociceptive and Anti-inflammatory Conjugate of Naturally Occurring Oleanolic Acid and Aspirin. Front. Pharmacol. 2016, 7, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Kleszcz, R.; Baer-Dubowska, W. The Effect of Novel Oleanolic Acid Oximes Conjugated with Indomethacin on the Nrf2-ARE And NF-κB Signaling Pathways in Normal Hepatocytes and Human Hepatocellular Cancer Cells. Pharmaceuticals 2020, 14, 32. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Kim, S.; Lee, Y.; Lee, H.; Lee, Y.; Park, H.; Nahm, J.H.; Ahn, S.; Yu, S.J.; Lee, K.; et al. The clinicopathological and prognostic significance of nrf2 and keap1 expression in hepatocellular carcinoma. Cancers 2020, 12, 2128. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Shi, F.; Sun, G.C.; Chen, H. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem. Biol. Interact. 2013, 206, 100–108. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baer-Dubowska, W.; Narożna, M.; Krajka-Kuźniak, V. Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs. Molecules 2021, 26, 4957. https://doi.org/10.3390/molecules26164957
Baer-Dubowska W, Narożna M, Krajka-Kuźniak V. Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs. Molecules. 2021; 26(16):4957. https://doi.org/10.3390/molecules26164957
Chicago/Turabian StyleBaer-Dubowska, Wanda, Maria Narożna, and Violetta Krajka-Kuźniak. 2021. "Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs" Molecules 26, no. 16: 4957. https://doi.org/10.3390/molecules26164957
APA StyleBaer-Dubowska, W., Narożna, M., & Krajka-Kuźniak, V. (2021). Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs. Molecules, 26(16), 4957. https://doi.org/10.3390/molecules26164957