
On the Origins of Some Spectroscopic Properties of “Purple Iron” (the Tetraoxoferrate(VI) Ion) and Its Pourbaix Safe-Space



Abstract
:1. Introduction
1.1. Examples of the Latest Applications of Ferrate(VI)
1.2. Chemical Feasibility as a Consideration in the Choice of Oxidants
1.3. Interesting Case Studies in Instrumental Methods of Analysis for Ferrate(VI)
2. Literature Survey
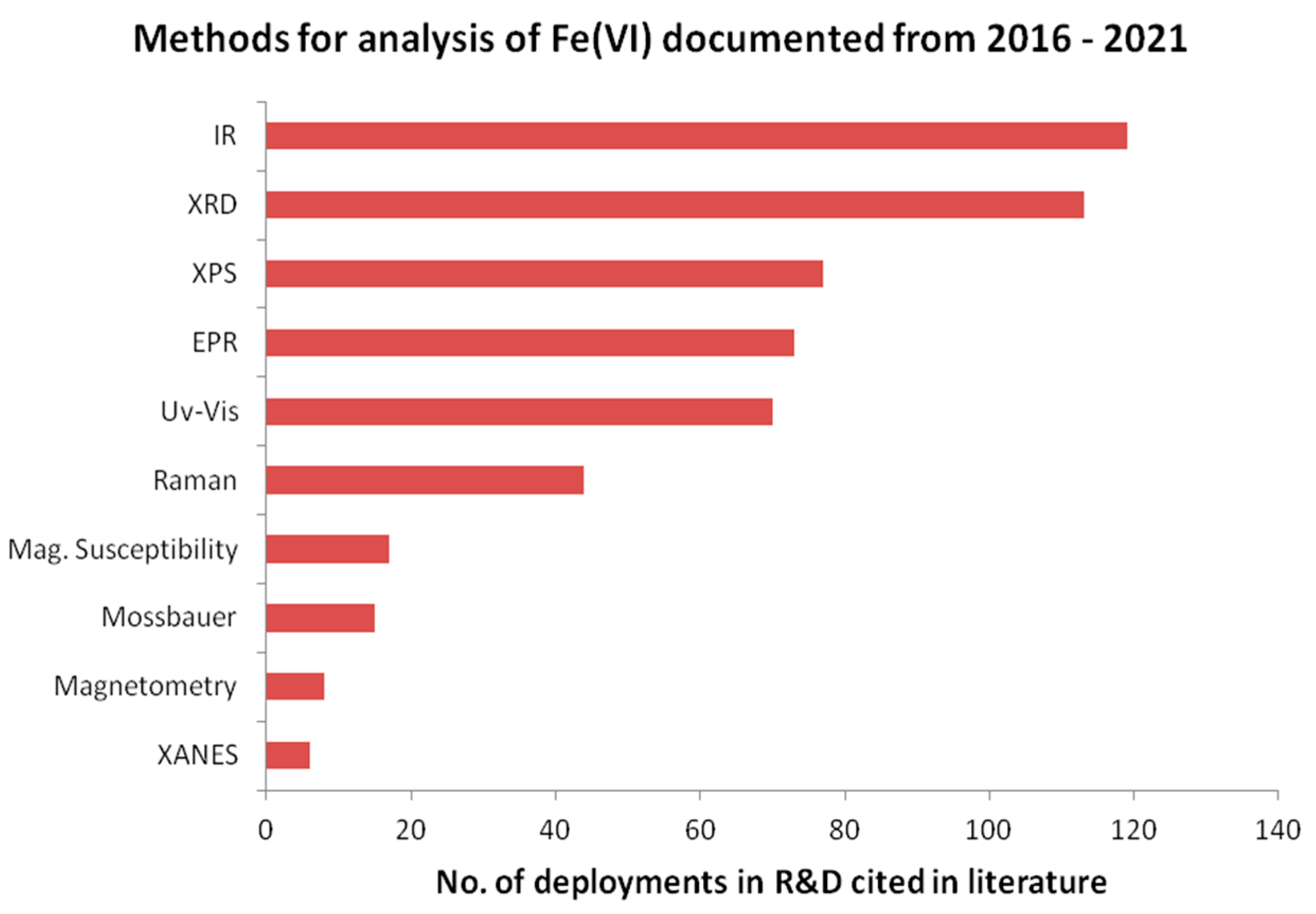
2.1. Instrumental Methods of Analyses Available to Research Activities
- Nanoparticles and nanostructures.
- Biological interactions of silver and gold nanoparticles.
- Water treatment and Photocatalysis.
- Examination of crystals, complexes and derivatives.
2.2. The Tetraoxoferrate (VI) Ion
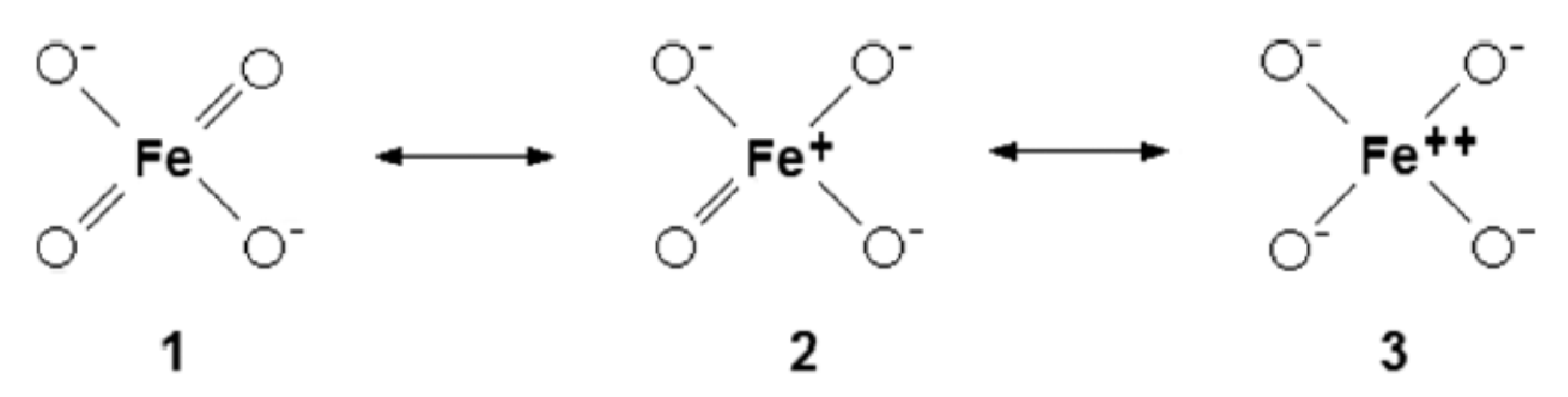
3. Molecular Orbital Theory Applied to Ferrate(VI)
Symmetry and Bonding in Ferrate(VI)
- 1st row elements: V(+V)O43−, Cr(+VI)O42− and Mn(+VII)O4−. Note that, in aqueous solutions, MnVO43− and MnVIO42− are extremely unstable at any pH. Both disproportionate into MnVIIO4−, and to Mn2+ (in acidic solution), or to MnIVO2 (in alkaline solution).
- 2nd row elements: [Mo(+VI)O4]2−, [Tc(+VII)O4]−, Ru(+VIII)O4.
- 3rd row elements: [W(+VI)O4]2−, [Re(+VII)O4]−, Os(+VIII)O4.
4. Electronic Spectra
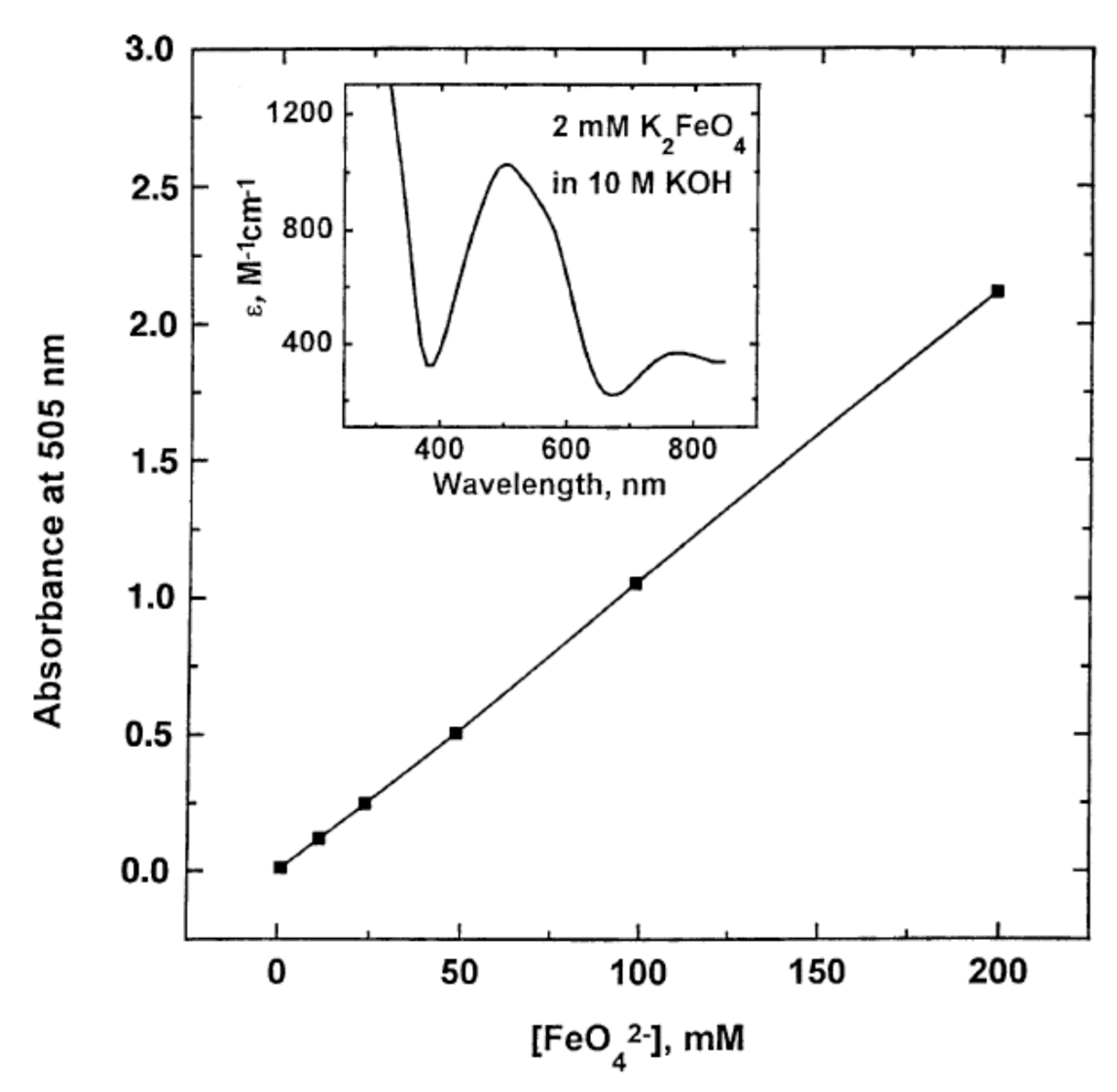
4.1. The Absorption Spectrum of Ferrate(VI)
4.2. The Orgel Diagram for Tetrahedral Complexes
5. Vibrational Spectra
5.1. Normal Modes and Fundamental Bands
5.2. Number of Vibrational Modes
5.3. Degenerate Vibrations in Point Group Td
5.4. Infrared and Raman Activities
5.5. History of Analysis of Ferrate (VI) by Vibration Spectroscopy
5.6. Td Symmetry of FeO42− Preserved under Highly Alkaline Conditions
5.7. The Search for the “FeO4” Molecule: Implications for Modelling [FeVIO4]2−
6. Stability and Solubility of Iron Species in Aqueous Solutions
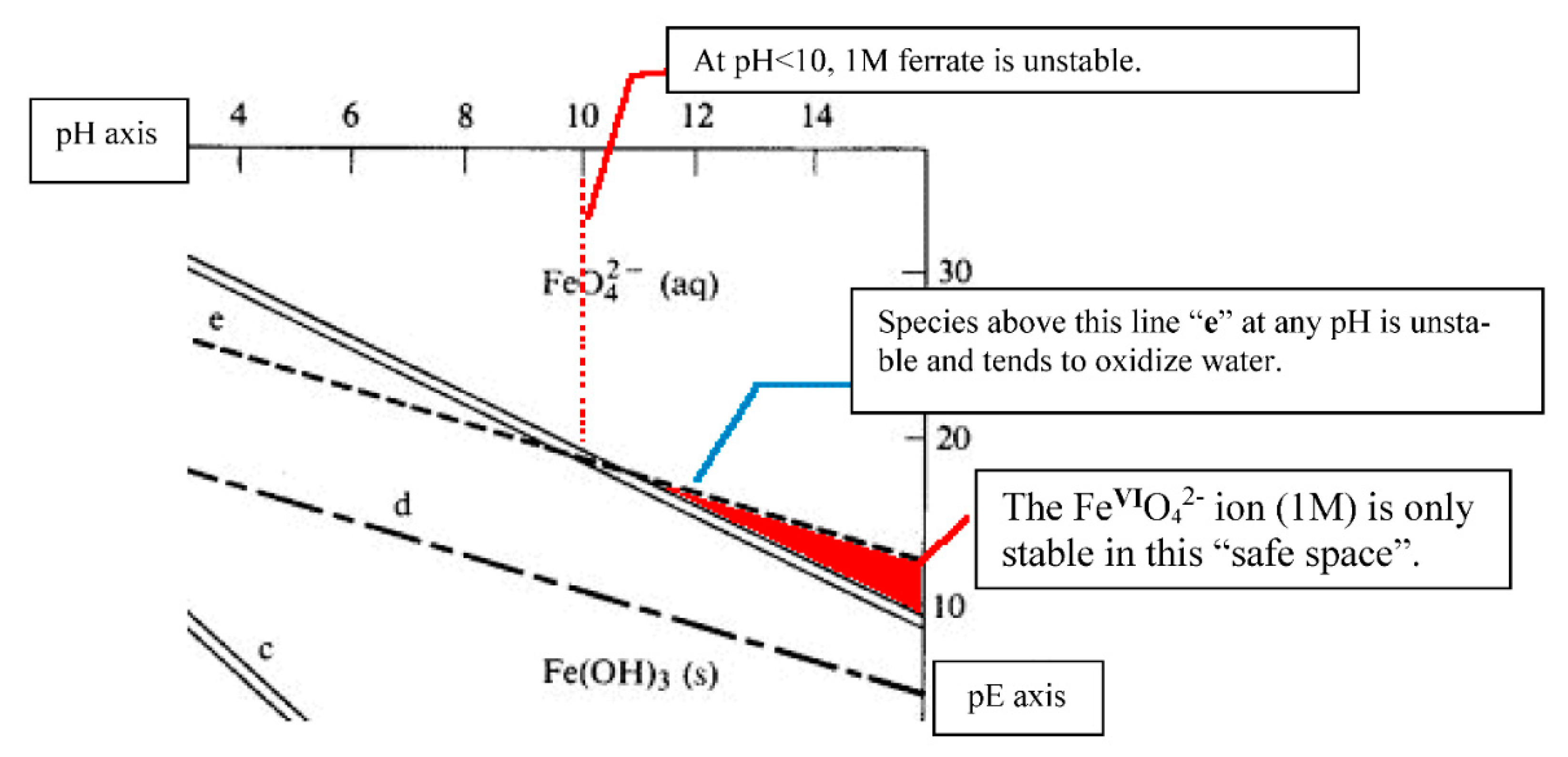
6.1. Considerations of Pourbaix Stability
6.2. The Sacrificial Decomposition of Tetraoxoferrate(VI)
6.2.1. The Quest for the Optimal pH as an Operational Parameter in Water Treatment
6.2.2. The Pioneer Work of Goff & Murmann
6.2.3. The Role of Group II Metal Ions in Ferrate(VI) Decomposition in Alkaline Waters
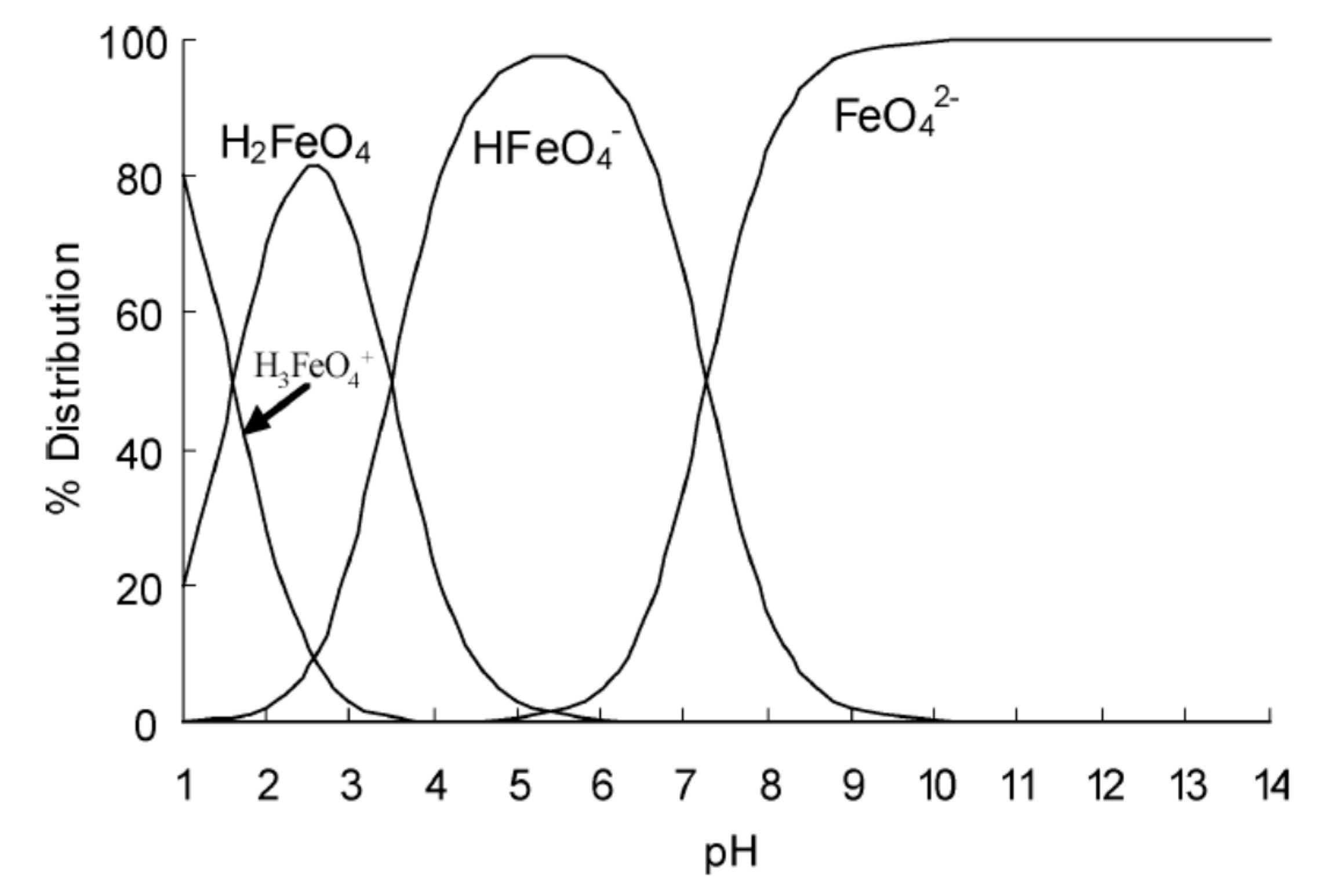
6.2.4. Protonated Forms of Ferrate(VI)
6.2.5. Decomposition of Ferrate(VI) in Acidic Solutions
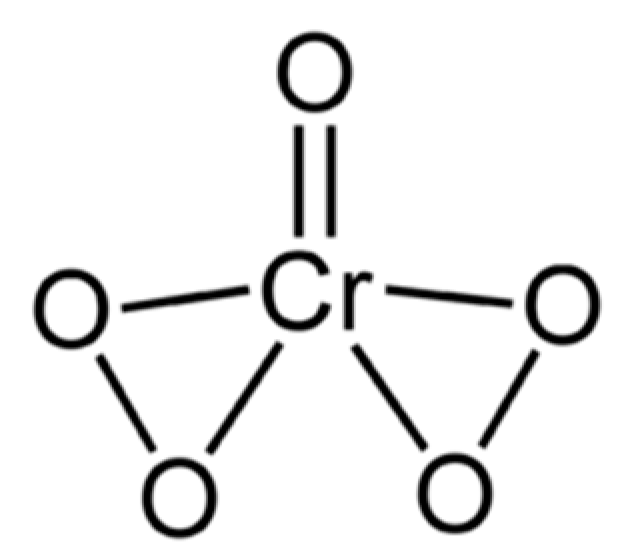
6.2.6. The Dichromate Analogy
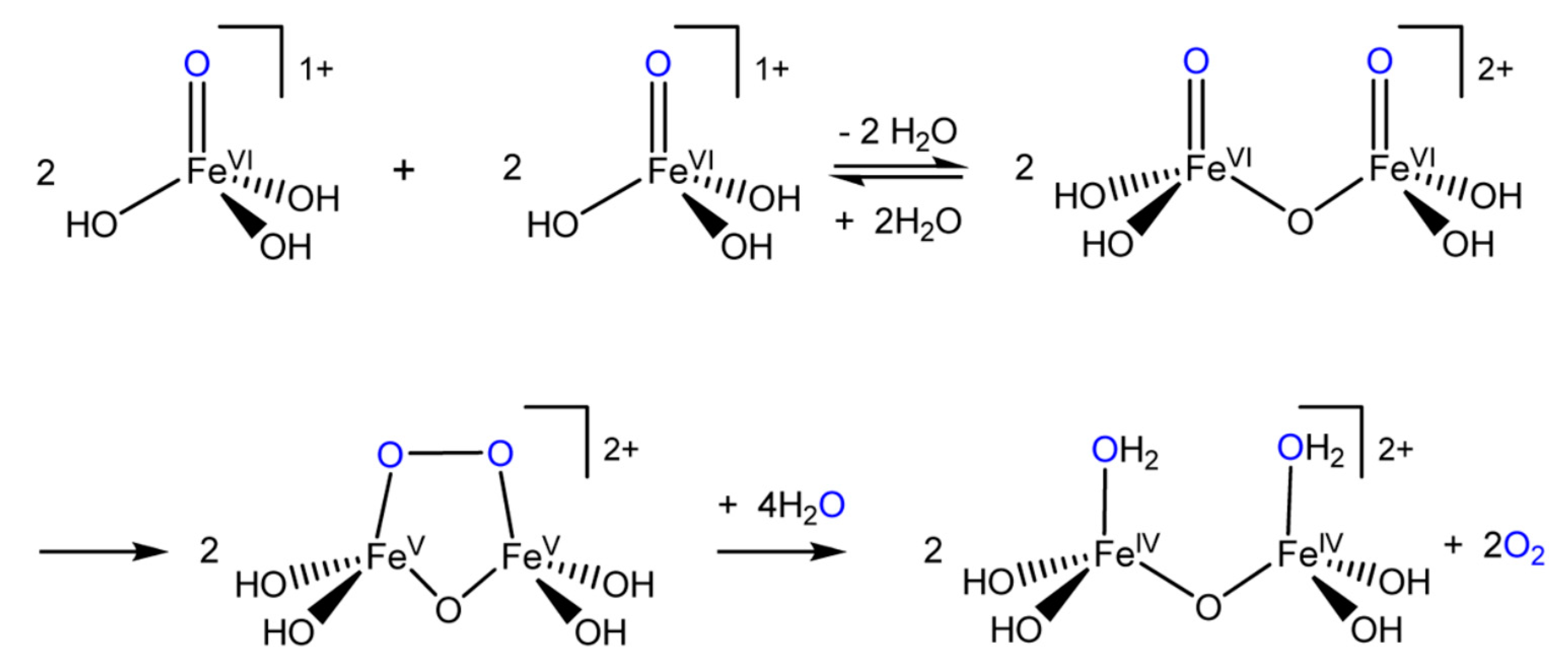
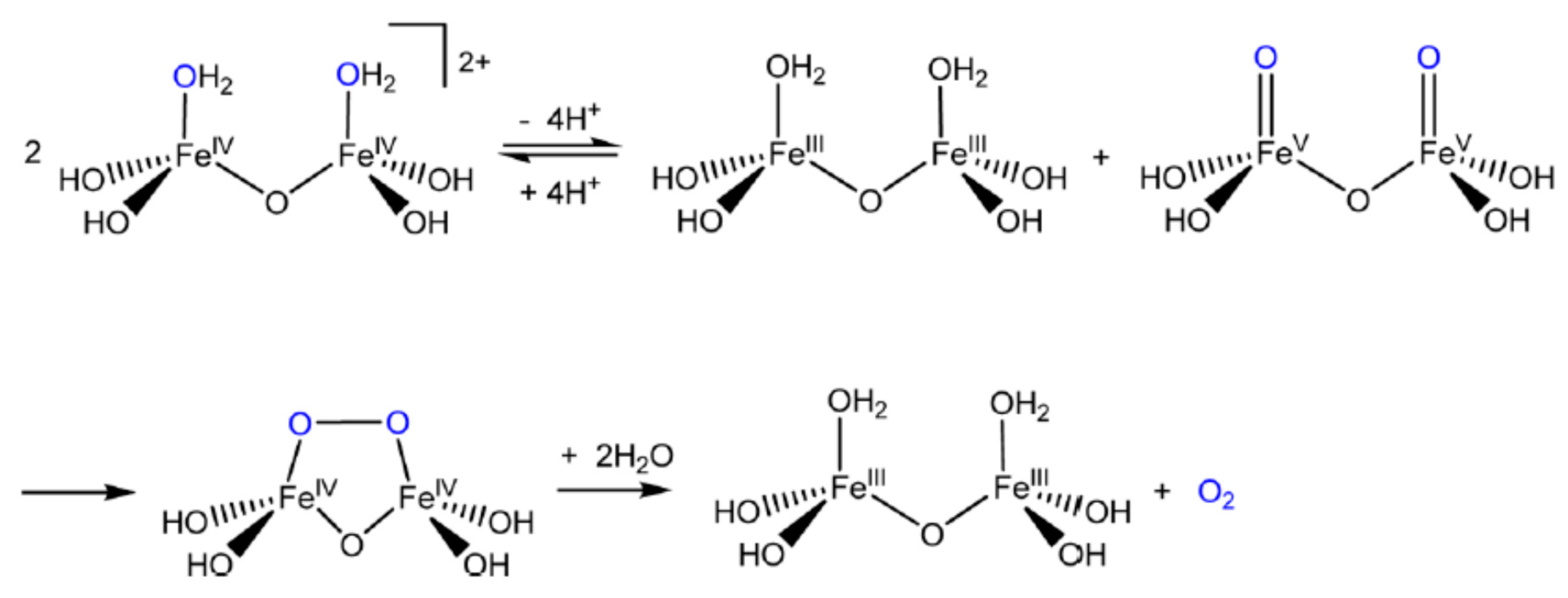
6.2.7. Descriptions of Some Proposed Reaction Pathways
6.2.8. The Dichromate Analogue Incorporated into Mathematical Formulation
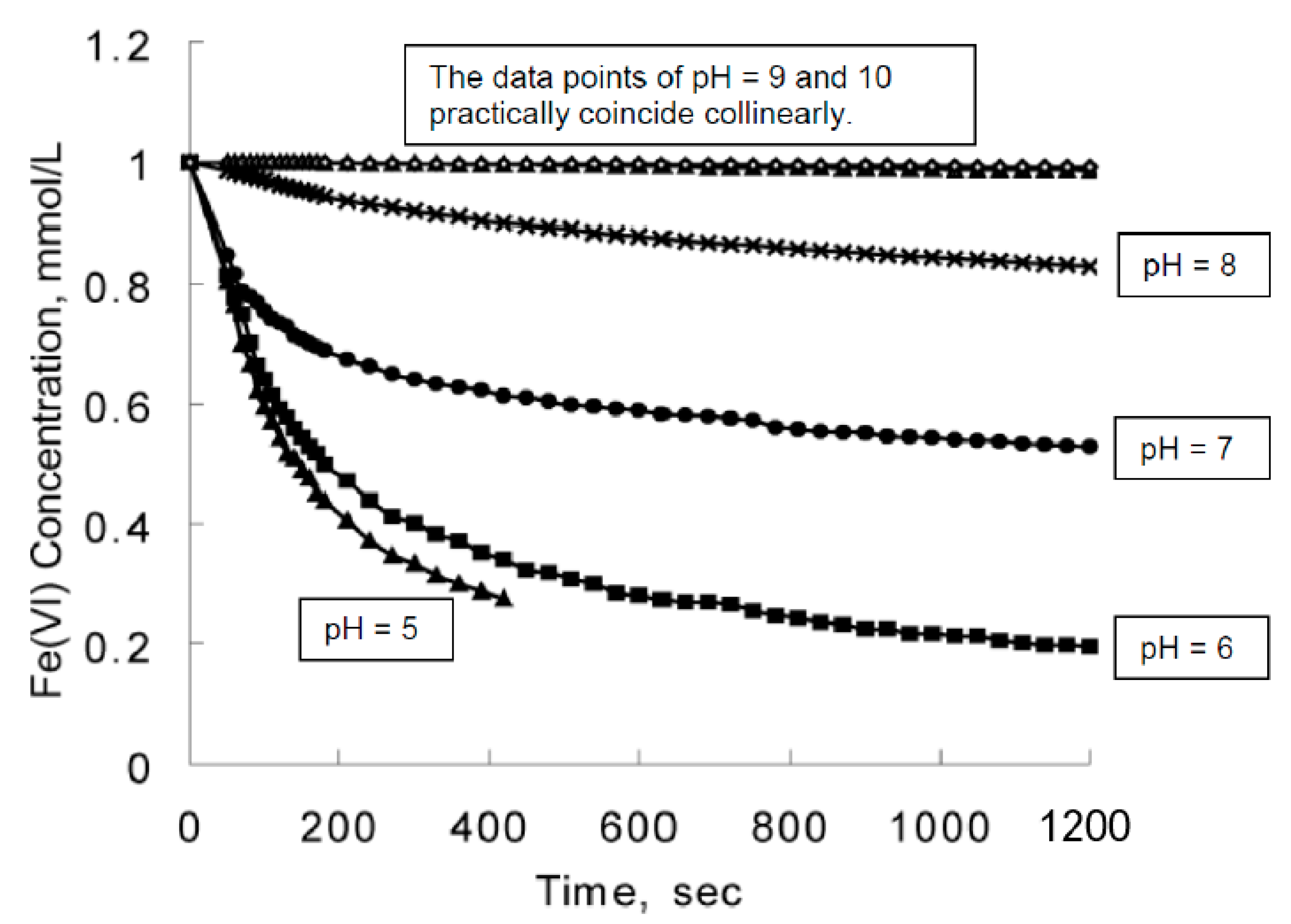
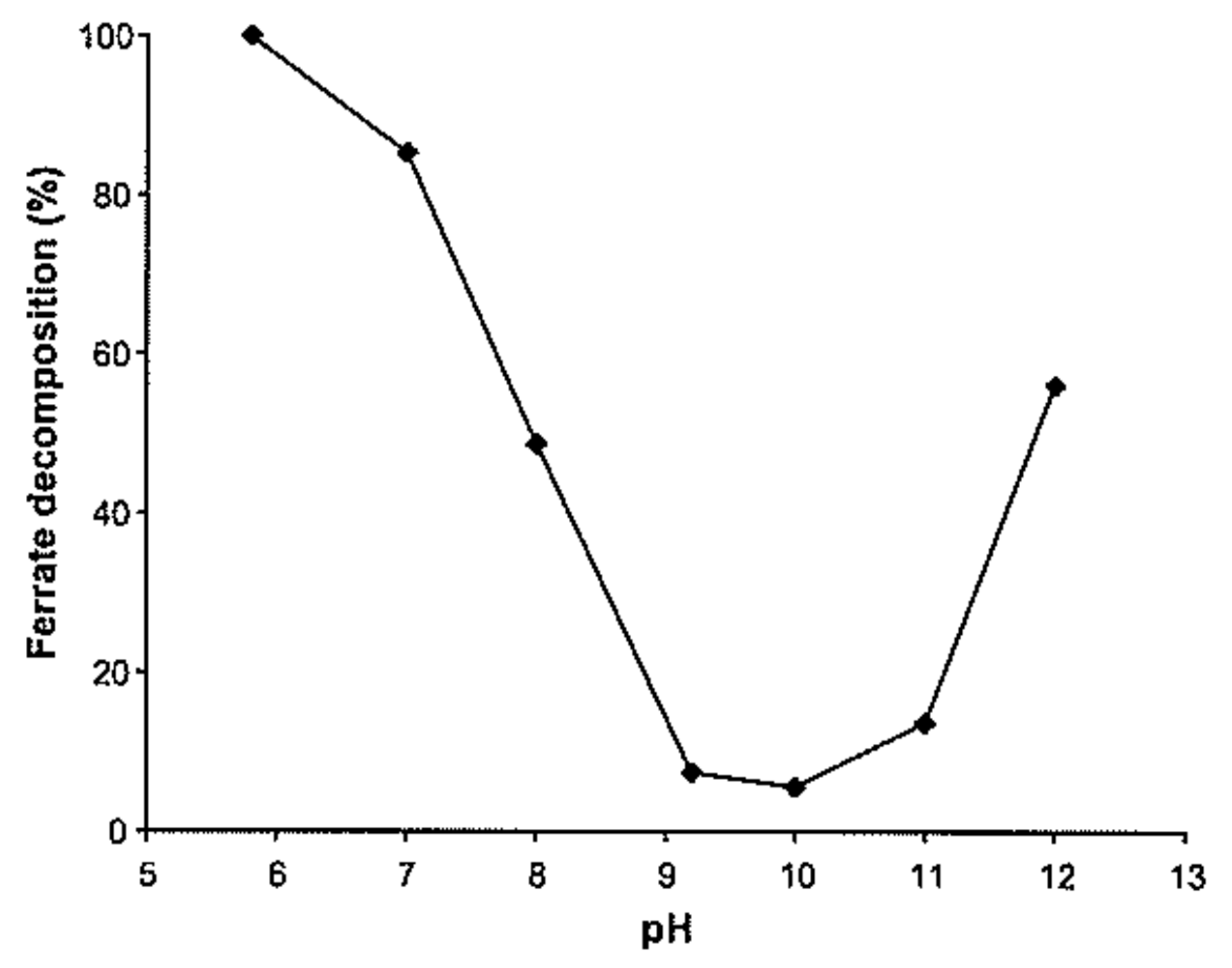
6.2.9. Strong Dependency of Ferrate(VI) Decomposition on pH Observed in Laboratories
6.3. A Self-Imposed Limitation of Fe(VI) Redox Reactions
6.4. The Solubility Products of Fe(II) and Fe(III) Hydroxides
7. Further Work
7.1. Acquire the Inelastic Neutron Scattering (INS) Spectra of Ferrate(VI)
7.2. Re-Investigate the Rate of Ferrate(VI) Self-Decay from Mild to High Alkalinity
- (a)
- pH = 9, 10, 11, 12, 13 and 14.
- (b)
- Concentrations of ferrate = 1, 5, 10 and 20, 50 mM of analytical grade K2FeO4.
- (c)
- Temperatures of aqueous solutions = 5 °C, 15 °C, 25 °C, 35 °C.
7.3. Investigation of the Effect of the Metal Ions on Ferrate(VI) Decomposition
7.4. Investigation of Components of Iron Oxide-Hydroxide Sludges as Part of the Research
7.5. Storage of Solid K2FeO4
7.6. Further Reading: Latest Theoretical Developments on Chemical Bonding
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diaz, M.; Doederer, K.; Keller, J.; Cataldo, M.; Donose, B.C.; Ali, Y.; Ledezma, P. Towards in situ electro-generation of ferrate for drinking water treatment: A comparison of three low-cost sacrificial iron electrodes. J. Electroanal. Chem. 2021, 880, 114897. [Google Scholar] [CrossRef]
- Licht, S. Measurements of pH in concentrated alkaline solutions. Anal. Chem. 1985, 57, 514–519. [Google Scholar] [CrossRef]
- McBeath, S.T.; Graham, N.J.D. Simultaneous electrochemical oxidation and ferrate generation for the treatment of atrazine: A novel process for water treatment applications. J. Hazard. Mater. 2021, 411, 125167. [Google Scholar] [CrossRef]
- Munyengabe, A.; Zvinowanda, C.; Zvimba, J.N.; James Ramontja, J. Characterization and reusability suggestions of the sludge generated from a synthetic acid mine drainage treatment using sodium ferrate(VI). Heliyon 2020, 6, e05244. [Google Scholar] [CrossRef] [PubMed]
- Alshahri, A.H.; Fortunato, L.; Ghaffour, N.; Leiknes, T. Advanced coagulation using in-situ generated liquid ferrate, Fe (VI), for enhanced pretreatment in seawater RO desalination during algal blooms. Sci. Total Environ. 2019, 685, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Addison, E.L.; Gerlach, K.T.; Spellman, C.D., Jr.; Santilli, G.; Fairbrother, A.R.; Shepard, Z.; Dudle, J.D.; Goodwill, J.E. Physicochemical implications of cyanobacteria oxidation with Fe(VI). Chemosphere 2020, 266, 128956. [Google Scholar] [CrossRef]
- Nikolić Bujanović, L.; Čekerevac, M.; Simičić, M.; Tomić, M. Encapsulation of micro-sized barium ferrate(VI) and its effectiveness in removing clomazone pesticide from water. J. Mater. Sci. 2020, 55, 7295–7303. [Google Scholar] [CrossRef]
- Acosta-Rangel, A.; Sánchez-Polo, M.; Rozalen, M.; Rivera-Utrilla, J.; Polo, A.M.S.; Berber-Mendoza, M.S.; López-Ramón, M.V. Oxidation of sulfonamides by ferrate(VI): Reaction kinetics, transformation byproducts and toxicity assesment. J. Environ. Manag. 2019, 255, 109927. [Google Scholar] [CrossRef] [PubMed]
- Monfort, O.; Usman, M.; Hanna, K. Ferrate(VI) oxidation of pentachlorophenol in water and soil. Chemosphere 2020, 253, 126550. [Google Scholar] [CrossRef]
- Cheung, P.C.W.; Williams, D.R. Separation of transition metals and chelated complexes in wastewaters. Environ. Prog. Sustain. Energy 2014, 34, 761–783. [Google Scholar] [CrossRef]
- Bryan, M.H.; Jay, R.W.; Harry, B.G. Iron Is the Active Site in Nickel/Iron Water Oxidation Electrocatalysts. Molecules 2018, 23, 903. [Google Scholar]
- Hunter, B.M.; Thompson, N.B.; Müller, A.M.; Rossman, G.R.; Hill, M.G.; Winkler, J.R.; Gray, H.B. Trapping an Iron(VI) Water-Splitting Intermediate in Nonaqueous Media. Joule 2018, 2, 747–763. [Google Scholar] [CrossRef] [Green Version]
- Alex, S. Johnson Matthey: Racing to survive. Chem. Eng. News 2021, 99, 22–23. [Google Scholar] [CrossRef]
- Glaze, W.H.; Kang, J.-W. Advanced Oxidation Processes for Treating Groundwater Contaminated with TCE and PCE: Laboratory Studies. J. Am. Water Work. Assoc. 1988, 80, 57–63. [Google Scholar] [CrossRef]
- Buffle, M.-O.; Schumacher, J.; Meylan, S.; Jekel, M.; von Gunten, U. Ozonation and Advanced Oxidation of Wastewater: Effect of O3 Dose, pH, DOM and HO•-Scavengers on Ozone Decomposition and HO•Generation. Ozone Sci. Eng. 2006, 28, 247–259. [Google Scholar] [CrossRef]
- Weeks, J.L.; Rabani, J. The Pulse Radiolysis of Deaerated Aqueous Carbonate Solutions. I. Transient Optical Spectrum and Mechanism. II. pK for OH Radicals1. J. Phys. Chem. 1966, 70, 2100–2106. [Google Scholar] [CrossRef]
- Dorfman, L.M.; Adams, G.E. Chapter 9: Reactions of the oxide radical ion. In Reactivity of the Hydroxyl Radical in Aqueous Solution; NSRDS-NBS 46; U.S. Department of Commerce: Washington, DC, USA, 1973; pp. 40–42. [Google Scholar]
- Lee, J.; Grabowski, J.J. Reactions of the atomic oxygen radical anion and the synthesis of organic reactive intermediates. Chem. Rev. 1992, 92, 1611–1647. [Google Scholar] [CrossRef]
- Buda, F.; Ensing, B.; Gribnau, M.C.; Baerends, E.J. DFT study of the active intermediate in Fenton reaction. Chem. Eur. J. 2001, 7, 2775–2783. [Google Scholar] [CrossRef]
- Ensing, B.; Buda, F.; Baerends, E.J. Fenton-like chemistry in water: Oxidation catalysis by Fe(III) and H2O2. J. Phys. Chem. A 2003, 107, 5722–5731. [Google Scholar] [CrossRef]
- Tsapin, A.I.; Goldfeld, M.G.; McDonald, G.D.; Nealson, K.H.; Moskovitz, B.; Solheid, P.; Kemner, K.M.; Kelly, S.D.; Orlandini, K.A. Iron(VI): Hypothetical Candidate for the Martian Oxidant. Icarus 2000, 147, 68–78. [Google Scholar] [CrossRef]
- Crawford, R.L.; Paszczynski, A.; Allenbach, L. Potassium ferrate Fe(VI) does not mediate self-sterilization of a surrogate mars soil. BMC Microbiol. 2003, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, G.V. Iron(VI) Seems an Unlikely Explanation for Viking Labeled Release Results. Icarus 2002, 159, 266–267. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Halperin, L.; Halperin, N.; Lin, L.; Chen, J.; Ghosh, S.; Liu, B. Analysis of ferrate(VI) compounds and super-iron Fe(VI) battery cathodes: FTIR, ICP, titrimetric, XRD, UV/VIS, and electrochemical characterization. J. Power Sources 2001, 101, 167–176. [Google Scholar] [CrossRef]
- Sparks, D.L.; Brown, A.; Hermandez, R.; Benson, T.J.; Holmes, W.E. Analytical Chemistry for Chemical Engineers. Conference paper 478C. In Proceedings of the AIChE Annual Meeting, Nashville, TN, USA, 11 November 2009; Available online: https://www.researchgate.net/publication/267318202_Analytical_Chemistry_for_Chemical_Engineers (accessed on 1 July 2021).
- Experimental Methods in Chemical Engineering; Newsletter; John Wiley & Sons: New Jersey, NJ, USA, 2020; Available online: https://onlinelibrary.wiley.com/doi/toc/10.1002/(ISSN)1939-019x.experimental-methods (accessed on 1 July 2021).
- Rocha, F.S.; Gomes, A.J.; Lunardi, C.N.; Kaliaguine, S.; Patience, G.S. Experimental methods in chemical engineering: Ultraviolet visible spectroscopy-UV-Vis. Can. J. Chem. Eng. 2018, 96, 2512–2517. [Google Scholar] [CrossRef]
- Guerrero-Pérez, M.O.; Patience, G.S. Experimental methods in chemical engineering: Fourier transform infrared spectroscopy—FTIR. Can. J. Chem. Eng. 2019, 98, 25–33. [Google Scholar] [CrossRef]
- Guerrero-Pérez, M.O.; Patience, G.S.; Bańares, M.A. Experimental methods in chemical engineering: Raman spectroscopy. Can. J. Chem. Eng. 2020, 99, 97–107. [Google Scholar] [CrossRef]
- Bakkar, M.G.; Fowler, B.; Bowman, M.K.; Patience, G.S. Experimental methods in chemical engineering: Electron paramagnetic resonance spectroscopy (EPR/ESR). Can. J. Chem. Eng. 2020, 98, 1668–1681. [Google Scholar] [CrossRef]
- Gomes, A.J.; Lunardi, C.N.; Rocha, F.S.; Patience, G.S. Experimental methods in chemical engineering: Fluorescence emission spectroscopy. Can. J. Chem. Eng. 2019, 97, 2168–2175. [Google Scholar] [CrossRef]
- Luo, Z.Y.; Strouse, M.; Jiang, J.Q.; Sharma, V.K. Methodologies for the analytical determination of ferrate(VI): A Review. J. Environ. Sci. Health Part A 2011, 46, 453–460. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G. Chapter 21: The elements of the first transition series. In Advanced Inorganic Chemistry, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1980; p. 765. [Google Scholar]
- Šišková, K.M.; Jančula, D.; Drahoš, B.; Machala, L.; Babica, P.; Alonso, P.G.; Trávníček, Z.; Tuček, J.; Maršálek, B.; Sharma, V.K.; et al. High-valent iron (Fe(VI), Fe(V), and Fe(IV)) species in water: Characterization and oxidative transformation of estrogenic hormones. Phys. Chem. Chem. Phys. 2016, 18, 18802–18810. [Google Scholar] [CrossRef] [Green Version]
- Machala, L.; Zboril, R.; Sharma, V.K.; Filip, J.; Schneeweiss, O.; Homonnay, Z. Mössbauer Characterization and in situ Monitoring of Thermal Decomposition of Potassium Ferrate(VI), K2FeO4 in Static Air Conditions. J. Phys. Chem. B 2007, 111, 4280–4286. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Homonnay, Z.; Siskova, K.; Machala, L.; Zboril, R. Mössbauer investigation of the reaction of ferrate(VI) with sulfamethoxazole and aniline in alkaline medium. Hyperfine Interact. 2014, 224, 7–13. [Google Scholar] [CrossRef]
- Virender, K.S.; Zboril, R.; Rajender, S.V. Ferrates(IV, V, and VI): Mössbauer Spectroscopy Characterization, Mössbauer Spectroscopy; Chapter, 24, Sharma, V.K., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 505–520. [Google Scholar]
- Autschbach, J.; Ziegler, T. Double perturbation theory: A powerful tool in computational coordination chemistry. Coord. Chem. Rev. 2003, 238–239, 83–126. [Google Scholar] [CrossRef]
- Schmidbaur, H. The History and the Current Revival of the Oxo Chemistry of Iron in its Highest Oxidation States: FeVI–FeVIII. Z. Für Anorg. Allg. Chem. 2018, 644, 536–559. [Google Scholar] [CrossRef]
- Carrington, A.; Ingram, D.J.E.; Schonland, D.; Symons, M.C.R. Structure and reactivity of the oxy-anions of transition metals. Part II. Investigations by electron-spin resonance. J. Chem. Soc. 1956, 1956, 4710. [Google Scholar] [CrossRef]
- Carrington, A.; Ingram, D.J.E.; Lott, K.A.K.; Schonland, D.S.; Symons, M.C.R. Electron Resonance Studies of Transition Metal Oxyions. I. Experimental Results for the Manganate, Hypomanganate and Ferrate Ions. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1960, 254, 101–110. [Google Scholar]
- Krebs, H. Über die Struktur des Kaliumferrats und des Bariumferrats Mit 2 Abbildungen. Z. Für Anorg. Allg. Chem. 1950, 263, 175–176. [Google Scholar] [CrossRef]
- Scholder, R. Zur Kenntnis der Ferrate(VI). Z. Für Anorg. Allg. Chem. 1955, 282, 268–279. [Google Scholar] [CrossRef]
- Griffith, W.P. Infrared spectra of tetrahedral oxyanions of the transition metals. J. Chem. Soc. A 1966, 1467–1468. [Google Scholar] [CrossRef]
- Goff, H.; Murmann, R.K. Mechanism of isotopic oxygen exchange and reduction of ferrate(VI) ion (FeO42−). J. Am. Chem. Soc. 1971, 93, 6058–6065. [Google Scholar] [CrossRef]
- Ziegler, T.; Li, J. Density Functional Study of C-H and O-H Activation and Methanol Oxidation by Chromyl Chloride. Organometallics 1995, 14, 214–223. [Google Scholar] [CrossRef]
- Norcross, B.E.; Lewis, W.C.; Gai, H.; Noureldin, N.A.; Lee, D.G. The oxidation of secondary alcohols by potassium tetraoxoferrate(VI). Can. J. Chem. 1997, 75, 129–139. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A.; Baerends, E. The electronic structures of tetrahedral oxo-complexes. The nature of the “charge transfer” transitions. Chem. Phys. 1976, 16, 209–217. [Google Scholar] [CrossRef]
- Jitsuhiro, S.; Nakai, H.; Hada, M.; Nakatsuji, H. Theoretical study on the ground and excited states of the chromate anion [CrO4]2−. J. Chem. Phys. 1994, 101, 1029–1036. [Google Scholar] [CrossRef]
- Ghernaout, D.; Naceur, M. Ferrate(VI): In situ generation and water treatment–A review. Desalin. Water Treat. 2011, 30, 319–332. [Google Scholar] [CrossRef]
- Talaiekhozani, A.; Bagheri, M.; Khozani, M.R.T.; Jaafarzadeh, N. An Overview on Production and Applications of Ferrate(VI). Jundishapur J. Health Sci. 2016, 8, e34904. [Google Scholar] [CrossRef] [Green Version]
- Burgess, J. Chapter 4: Ion-solvent interactions. In Ions in Solution; Burgess, J., Ed.; Woodhead Publishing: Cambridge, UK, 1999; pp. 45–61. [Google Scholar]
- Schröder, D.; Fiedler, A.; Schwarz, J.; Schwarz, H. Generation and Characterization of the Anionic, Neutral, and Cationic Iron-Dioxygen Adducts [FeO2] in the Gas Phase. Inorg. Chem. 1994, 33, 5094–5100. [Google Scholar] [CrossRef]
- Wu, H.; Desai, S.R.; Wang, L. Observation and Photoelectron Spectroscopic Study of Novel Mono- and Diiron Oxide Molecules. J. Am. Chem. Soc. 1996, 118, 5296–5301. [Google Scholar] [CrossRef]
- Jose, L.; Seth, M.; Ziegler, T. Molecular and Vibrational Structure of Tetroxo d0 Metal Complexes in their Excited States. A Study Based on Time-Dependent Density Functional Calculations and Franck–Condon Theory. J. Phys. Chem. A 2012, 116, 1864–1876. [Google Scholar] [CrossRef]
- Seth, M.; Mazur, G.; Ziegler, T. Time-dependent density functional theory gradients in the Amsterdam density functional package: Geometry optimizations of spin-flip excitations. Theor. Chem. Acc. 2011, 129, 331–342. [Google Scholar] [CrossRef]
- Stückl, A.C.; Daul, C.A.; Güdel, H.U. Excited-state energies and distortions of d0 transition metal tetraoxo complexes: A density functional study. J. Chem. Phys. 1997, 107, 4606–4617. [Google Scholar] [CrossRef]
- Neugebauer, J.; Baerends, E.J.; Nooijen, M. Vibronic Structure of the Permanganate Absorption Spectrum from Time-Dependent Density Functional Calculations. J. Phys. Chem. A 2005, 109, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Seidu, I.; Krykunov, M.; Ziegler, T. Applications of Time-Dependent and Time-Independent Density Functional Theory to Electronic Transitions in Tetrahedral d0 Metal Oxides. J. Chem. Theory Comput. 2015, 11, 4041–4053. [Google Scholar] [CrossRef]
- Reinen, D.; Rauwa, W.; Kespera, U.; Atanasovb, M.; Güdel, H.U.; Hazenkamp, M.; Oetliker, U. Colour, luminescence and bonding properties of tetrahedrally coordinated chromium(IV), manganese(V) and iron(VI) in various oxide ceramics. J. Alloy. Compd. 1997, 246, 193–208. [Google Scholar] [CrossRef]
- Berry, J.F.; Bill, E.; Bothe, E.; George, S.D.; Mienert, B.; Neese, F.; Wieghardt, K. An Octahedral Coordination Complex of Iron(VI). Science 2006, 312, 1937–1941. [Google Scholar] [CrossRef] [Green Version]
- di Sipio, L.; de Michelis, G.; Baiocchi, E.; Ingletto, G. Electronic Adsorption Spectra of Tetrahedral Oxoanions: Ferrate(VI) Ion. Trans. Met. Chem. 1980, 5, 164–167. [Google Scholar] [CrossRef]
- Purcell, K.F.; Kotz, J.C. Fundamental Concepts for Transition Metal Complexes. In Inorganic Chemistry; Chapter 9; W.B. Saunders: Philadelphia, PA, USA, 1977; p. 543. [Google Scholar]
- Carrington, A.; Schonland, D.; Symons, M. Structure and reactivity of the oxyanions of transition metals. Part IV. Some relations between electronic spectra and structure. J. Chem. Soc. 1957, 659–665. [Google Scholar] [CrossRef]
- Al-Abdalla, A.; Seijo, L.; Barandiarán, Z. A new interpretation of the bonding and spectroscopy of the tetraoxoferrate(VI) FeO42− ion. J. Chem. Phys. 1998, 109, 6396–6405. [Google Scholar] [CrossRef] [Green Version]
- Guenzburger, D.; Darci, M.S. Esquivel; Jacques Danon. Electronic structure and hyperfine interactions in the FeO42− ion. Phys. Rev. B 1978, 18, 4561–4569. [Google Scholar] [CrossRef]
- Lever, A.B.P. Inorganic Electronic Spectra, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1984; p. 411. [Google Scholar]
- Wood, R.H. The Heat, Free Energy and Entropy of the Ferrate (VI) Ion. J. Am. Chem. Soc. 1958, 80, 2038–2041. [Google Scholar] [CrossRef]
- Kaufman, Z.G.; Schreyer, J.M. Spectrophotometry of the Ferrate(VI) Ion in Aqueous Solution. Chem. Anal. 1956, 45, 22–23. [Google Scholar]
- Carrington, A.; Symons, M.C.R. Structure and Reactivity of the Oxyanions of Transition Metals. Part IX. Electronic Spectra. J. Chem. Soc. 1960, 1, 889–891. [Google Scholar]
- Carrington, A.; Symons, M.C.R. Structure and Reactivity of the Oxyanions of Transition Metals (a review paper). Chem. Rev. 1963, 63, 443–460. [Google Scholar] [CrossRef]
- Barrett, J. Transition Metal Complexes. Chapter 8. In Understanding Inorganic Chemistry: The Underlying Physical Principles; Series in Inorganic Chemistry; Ellis Horwood Ltd.: Chichester, UK, 1991; p. 219. [Google Scholar]
- Sharma, V. Disinfection performance of Fe(VI) in water and wastewater: A review. Water Sci. Technol. 2007, 55, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Yoon, J.; von Gunten, U. Spectrophotometric determination of ferrate (Fe(VI)) in water by ABTS. Water Res. 2005, 39, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.W.Y.; Man, W.-L.; Wang, Y.-N.; Lau, T.-C. Kinetics and mechanisms of the oxidation of iodide and bromide in aqueous solutions by a trans-dioxoruthenium(VI) complex. Inorg. Chem. 2008, 47, 6771–6778. [Google Scholar] [CrossRef]
- Mulliken, R.S. Report on Notation for the Spectra of Polyatomic Molecules. J. Chem. Phys. 1955, 23, 1997–2011, Erratum in 1956, 24, 1118. [Google Scholar] [CrossRef]
- Schutte, C.J.H.; Bertie, J.E.; Bunker, P.R.; Hougen, J.T.; Mills, I.M.; Watson, J.K.G.; Winnewisser, B.P. Notations and Conventions in Molecular Spectroscopy: Part 2. Symmetry Notation: (IUPAC Recommendations 1997). In IUPAC Standards Online; De Gruyter: Berlin, Germany; Boston, MA, USA, 2016. [Google Scholar]
- Gonzalez-Vilchez, F.; Griffith, W.P. Transition-metal Tetra-oxo-complexes and their Vibrational Spectra. J. Chem. Soc. 1972, 13, 1416–1421. [Google Scholar] [CrossRef]
- Gray, D.; Robiette, A. The anharmonic force field and equilibrium structure of methane. Mol. Phys. 1979, 37, 1901–1920. [Google Scholar] [CrossRef]
- Walton, P.H. Beginning Group Theory for Chemistry; Chapter 6; Oxford University Press: Oxford, UK, 1998; pp. 79–111. [Google Scholar]
- Papanastasopoulos, C. Applications of Group Theory to the Vibrations of Methane Molecules in the Gaseous and Solid Phases. Master’s Thesis, Department of Physics, University of Montreal, Montreal, QC, Canada, 1972; pp. 5–24. [Google Scholar]
- Tuschel, D. Practical Group Theory and Raman Spectroscopy, Part II: Application of Polarization. Spectroscopy 2014, 29, 14–23. [Google Scholar]
- Becarud, N.; Dural, C. Reactions and properties of potassium, barium and strontium ferrates. Comptes Rendus 1963, 257, 1930–1933. [Google Scholar]
- Tarte, P.; Nizet, G. Etude infra-rouge de quelques composés du type K2SO4 et BaSO4. Spectrochim. Acta 1964, 20, 503–513. [Google Scholar] [CrossRef]
- Woodward, L.A.; Roberts, H.L. The Raman and infra-red absorption spectra of osmium tetroxide. Relation to the structure of the perrhenate and tungstate ions in aqueous solution. Trans. Faraday Soc. 1956, 52, 615–619. [Google Scholar] [CrossRef]
- El Maghraoui, A.; Zerouale, A.; Ijjaali, M. Effect of degree of hypochlorite on the wet synthesis of ferrate(VI). Adv. Mater. Phys. Chem. 2015, 5, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Brunold, T.C.; Güdel, H.; Kuck, S.; Huber, G. Excited state properties of ferrate(VI) doped crystals of K2SO4 and K2CrO4. J. Lumin. 1995, 65, 293–301. [Google Scholar] [CrossRef]
- Audette, R.J.; Quail, J.W.; Potassium, Rubidium, Cesium, and Barium Ferrates(VI). Preparations, Infrared Spectra, and Magnetic Susceptibilities. Inorg. Chem. 1972, 11, 1904–1908. [Google Scholar] [CrossRef]
- Sharma, V.K.; Burnett, C.R.; Millero, F.J. Dissociation constants of the monoprotic ferrate(VI) ion in NaCl media. Phys. Chem. Chem. Phys. 2001, 3, 2059–2062. [Google Scholar] [CrossRef]
- Schwaab, G.; Sebastiani, F.; Havenith, M. Ion Hydration and Ion Pairing as Probed by THz Spectroscopy. Angew. Chem. Int. Ed. 2019, 58, 3000–3013. [Google Scholar] [CrossRef]
- Schreyer, J.M.; Ockerman, L.T. Stability of Ferrate(VI) Ion in Aqueous Solution. Anal. Chem. 1951, 23, 1312–1314. [Google Scholar] [CrossRef]
- Carrington, A.; Symons, M.C.R. Structure and reactivity of the oxyanions of transition metals. Part VII. Kinetics and mechanism of the alkaline decomposition of per-ruthenate. J. Chem. Soc. 1960, 54, 284–289. [Google Scholar] [CrossRef]
- Griffith, W.P. Infrared spectra and structures of osmium oxy-complexes. J. Chem. Soc. 1964, 40, 245–249. [Google Scholar] [CrossRef]
- Luo, C.; Feng, M.; Sharma, V.K.; Huang, C.H. Revelation of ferrate(VI) unimolecular decay under alkaline conditions: Investigation of involvement of Fe(IV) and Fe(V) species. Chem. Eng. J. 2020, 388, 124134. [Google Scholar] [CrossRef]
- Griffith, W.P. Transition metal oxo complexes. Coord. Chem. Rev. 1970, 5, 459–517. [Google Scholar] [CrossRef]
- Goralevich, D.K. Higher oxygen compounds of elements of the eighth group of the periodic system of elements. I. Compounds of iron. J. Russ. Phys. Chem. Soc. 1926, 58, 1129–1158. [Google Scholar]
- Poleshchuk, O.; Kruchkova, N.; Perfiliev, Y.; Dedushenko, S. Estimations of the isomer shifts for tetraoxoferrates. J. Phys. Conf. Ser. 2010, 217, 12041. [Google Scholar] [CrossRef]
- Sharma, V.; Zboril, R. Ferryl and Ferrate Species: Mössbauer Spectroscopy Investigation. Croat. Chem. Acta 2015, 88, 363–368. [Google Scholar] [CrossRef]
- Kopelev, N.; Perfiliev, Y.; Kiselev, Y. Mössbauer spectroscopy of iron oxocompounds in higher oxidation states. J. Radioanal. Nucl. Chem. Artic. 1992, 157, 401–414. [Google Scholar] [CrossRef]
- Dasent, W.E. Non-Existent Compounds; Marcel Dekker: New York, NY, USA, 1965; p. 125. [Google Scholar]
- Dasent, W.E. Non-existent compounds. J. Chem. Educ. 1963, 40, 130–134. [Google Scholar] [CrossRef]
- Schmidbaur, H. Book Review: Nonexistent Compounds—Compounds of Low Stability, By, W. E. Dasent. Angew. Chem. Int. Ed. 1966, 5, 980. [Google Scholar] [CrossRef]
- Schmidbaur, H. Compounds of unknown structure. Organoberyllium Compounds. In Gmelin Handbook of Inorganic Chemistry; Kubny, A., Mirbach, M., Krüerke, U., Slawisch, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1987; Volume B-e/1/1. [Google Scholar]
- Huang, W.; Xu, W.H.; Schwarz, W.H.E.; Li, J. On the Highest Oxidation States of Metal Elements in MO4 Molecules (M = Fe, Ru, Os, Hs, Sm, and Pu). Inorg. Chem. 2016, 55, 4616–4625. [Google Scholar] [CrossRef]
- Schwarz, W.H.E. The Full Story of the Electron Configurations of the Transition Elements. J. Chem. Educ. 2010, 87, 444–448. [Google Scholar] [CrossRef]
- Philipsen, P.H.T.; Baerends, E.J. Cohesive energy of 3d transition metals: Density function theory atomic and bulk calculations. Phys. Rev. B 1996, 54, 5326–5333. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Xing, D.H.; Lu, J.B.; Schwarz, W.H.E.; Li, J. How Much Can Density Functional Approximations (DFA) Fail? The Extreme Case of the FeO4 Species. J. Chem. Theory Comput. 2016, 12, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.; Manzhos, S. On the Charge State of Titanium in Titanium Dioxide. J. Phys. Chem. Lett. 2017, 8, 1593–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.X.; Li, W.L.; Lu, J.B.; Bao, J.L.; Yu, H.S.; Truhlar, D.G.; Gibson, J.K.; Marçalo, J.; Zhou, M.; Riedel, S.; et al. On the Upper Limits of Oxidation States in Chemistry. Angew. Chem. Int. Ed. 2018, 57, 3242–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baerends, E.J. Speech by Prof. Baerends on acceptance of Doctor Honoris Causa at Universitat de Girona, Spain, 8 May 2019. Available online: http://iqcc.udg.edu/wordpress/2019/05/16/prof-evert-jan-baerends-doctor-honoris-causa/ (accessed on 1 July 2021).
- Ziegler, T.; Seth, M.; Krykunov, M.; Autschbach, J.; Wang, F. Is charge transfer transitions really too difficult for standard density functional or are they just a problem for time-dependent density functional theory based on a linear response approach? J. Mol. Struct. 2009, 914, 106–109. [Google Scholar] [CrossRef]
- Ziegler, T. A Chronicle about the Development of Electronic Structure Theories for Transition Metal Complexes. In Molecular Electronic Structures of Transition Metal Complexes II. Structure and Bonding; Mingos, D., Day, P., Dahl, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 143. [Google Scholar]
- Marten, A.B.; Baerends, E.J. Analysis of nondynamical correlation in the metal-ligand bond. Pauli repulsion and orbital localization in MnO4−. J. Phys. Chem. 1990, 93, 4129–4141. [Google Scholar]
- Schmidbaur, H.; Schwarz, W.H.E. Permanganyl Fluoride: A Brief History of the Molecule MnO3F and of Those Who Cared For It. Chem. Eur. J. 2021, 27, 6848–6859. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 1st ed.; Pergamon Press: London, UK, 1966; pp. 201, 206–207, 211. [Google Scholar]
- Cheung, P.C.W.; Williams, D.R.; Kirk, D.W. A novel application of Pourbaix diagrams for the ad hoc prediction of thermodynamic behaviour of plutonium in aquatic environments. Am. J. Environ. Eng. Sci. 2018, 5, 24–33. [Google Scholar]
- Wulfsberg, G. Inorganic Chemistry; University Science Books: Sausalito, CA, USA, 2000; p. 289. [Google Scholar]
- Graham, N.; Jiang, C.C.; Li, X.Z.; Jiang, J.Q.; Ma, J. The influence of pH on the degradation of phenol and chlorophenols by potassium ferrate. Chemosphere 2004, 56, 949–956. [Google Scholar] [CrossRef]
- Karim, A.V.; Krishnan, S.; Pisharody, L.; Malhotra, M. Application of Ferrate for Advanced Water and Wastewater Treatment; IntechOpen: London, UK, 2020; pp. 1–20. [Google Scholar]
- Jessen, A.; Randall, A.A.; Reinhart, D.R.; Daly, L. Effectiveness and kinetics of ferrate as a disinfectant for ballast water. Water Environ. Res. 2008, 80, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Western Oregon University. Pourbaix Diagrams. 2006. Available online: https://people.wou.edu/~courtna/ch412/pourbaix.htm (accessed on 1 July 2021).
- Chen, G.; Lam, W.W.; Lo, P.K.; Man, W.L.; Chen, L.; Lau, K.C.; Lau, T.C. Mechanism of Water Oxidation by Ferrate(VI) at pH 7–9. Chem. A Eur. J. 2018, 24, 18735–18742. [Google Scholar] [CrossRef]
- Rush, J.D.; Zhao, Z.; Bielski, B.H. Reaction of Ferrate (VI)/Ferrate (V) with Hydrogen Peroxide and Superoxide Anion—A Stopped-Flow and Premix Pulse Radiolysis Study. Free Radic. Res. 1996, 24, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, D.; Kim, H.U.; Choi, B.J.; Lee, S.M.; Kwon, O.H.; Choi, K.M.; Yang, J.K. Ferrate(VI): A green chemical for the oxidation of cyanide in aqueous/waste solutions. J. Environ. Sci. Health Part A 2007, 42, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, K. Chapter 5: Acids, bases and buffers. In Fundamentals of Biochemical Calculations, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2008; p. 56. [Google Scholar]
- Sarma, R.; Angeles-Boza, A.M.; Brinkley, D.W.; Roth, J.P. Studies of the Di-iron(VI) Intermediate in Ferrate-Dependent Oxygen Evolution from Water. J. Am. Chem. Soc. 2012, 134, 15371–15386. [Google Scholar] [CrossRef] [PubMed]
- European Chemicals Agency. Potassium Chromate. Registered Chemicals Information Sheet. 2005. Available online: https://www.echa.europa.eu/web/guest/registration-dossier/-/registered-dossier/5173/5/2/3 (accessed on 1 July 2021).
- Swineheart, J.H.; Castellan, W. The kinetics of the chromate-dichromate reaction as studied by a relaxation method. Inorg. Chem. 1964, 3, 278–280. [Google Scholar] [CrossRef]
- Holleman, A.F.; Wiberg, E.; Wiberg, N. Chromium. In Lehrbuch der Anorganischen Chemie, 91–100 ed.; Walter de Gruyter (Walter de Gruyter GmbH): Berlin, Germany, 1985; pp. 1081–1095. ISBN 3-11-007511-3. (In German). Available online: http://www.sciepub.com/reference/40795 (accessed on 1 July 2021).
- Bielski, B.H. Studies of Hypervalent Iron. Free Radic. Res. Commun. 1991, 13, 469–477. [Google Scholar] [CrossRef]
- Roth, J.P. Isotopic Studies of O-O Bond. Formation during Water Oxidation (SISGR); Final Technical Report for U.S. Department of Energy, Grant Number DE-SC0002508; U.S. Department of Energy, Office of Scientific and Technical Information: Washington, DC, USA, 2015; pp. 7–9.
- Angeles-Boza, A.M.; Roth, J.P. Oxygen Kinetic Isotope Effects upon Catalytic Water Oxidation by a Monomeric Ruthenium Complex. Inorg. Chem. 2012, 51, 4722–4729. [Google Scholar] [CrossRef]
- Lee, Y.; Kissner, R.; von Gunten, U. Reaction of Ferrate(VI) with ABTS and Self-Decay of Ferrate(VI): Kinetics and Mechanisms. Environ. Sci. Technol. 2014, 48, 5154–5162. [Google Scholar] [CrossRef]
- Carr, J.D.; Kelteraaa, P.B.; Tabatabai, A.; Spichal, D.; Erickson, J.; Mclaughlin, C.W. Water Chlorination: Chemistry, Environmental Impact, and Health Effects (bk); Jolley, R.L., Bull, R.J., Davis, W.P., Katz, S., Roberts, M.H., Jr., Jacobs, V.A., Eds.; Lewis Publishers Inc.: Chelsea, MI, USA, 1985; Volume 5, pp. 1285–1298. ISBN 0873710053. [Google Scholar]
- Carr, J.D. Kinetics and Product Identification of Oxidation by Ferrate(VI) of Water and Aqueous Nitrogen Containing Solutes, Ferrates; Chapter 13; American Chemical Society Symposium Series; American Chemical Society: Washington, DC, USA, 2008; Volume 985, pp. 189–196. Available online: https://pubs.acs.org/doi/abs/10.1021/bk-2008-0985.ch013 (accessed on 1 July 2021).
- Masterton, W.; Hurley, C. Chemistry and Principles and Reactions, 8th ed.; Chapter 20, Question 34; Cengage Learning: Independence, KY, USA, 2016; p. 524. [Google Scholar]
- Smith, J.A.; Metz, C.R. Spectrophotometry Determination of the Chromate-Dichromate Equilibrium Constant. J. Indiana Acad. Sci. 1970, 80, 159–162. [Google Scholar]
- Brito, F.; Ascanio, J.; Mateo, S.; Hernández, C.; Araujo, L.; Gili, P.; Martín-Zarza, P.; Domínguez, S.; Mederos, A. Equilibria of chromate(VI) species in acid medium and ab initio studies of these species. Polyhedron 1997, 16, 3835–3846. [Google Scholar] [CrossRef]
- Rush, J.D.; Bielski, B.H.J. Kinetics of ferrate(V) decay in aqueous solution. A pulse-radiolysis study. Inorg. Chem. 1989, 28, 3947–3951. [Google Scholar] [CrossRef]
- Rush, J.D.; Bielski, B.H.J. Decay of Ferrate(V) in Neutral and Acidic Solutions. A Premix Pulse Radiolysis Study. Inorg. Chem. 1994, 33, 5499–5502. [Google Scholar] [CrossRef]
- Carr, J.; Goncharova, I.; Golovko, D.; McLaughlin, C.; Golovko, I.; Erickson, J. Study of the oxidation kinetics of nitrite ions by potassium ferrate(VI). Eastern-Eur. J. Enterp. Technol. 2018, 3, 18–25. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Liu, B.; Ghosh, S.; Halperin, N.; Halperin, L.; Rosen, D. Chemical synthesis of battery grade super-iron barium and potassium Fe(VI) ferrate compounds. J. Power Sources 2001, 99, 7–14. [Google Scholar] [CrossRef]
- Machala, L.; Zboril, R.; Sharma, V.K.; Homonnay, Z. Decomposition of Potassium Ferrate(VI) and Potassium Ferrate(III): In-situ Mössbauer Spectroscopy Approach. AIP Conf. Proc. 2008, 1070, 114–121. [Google Scholar]
- Leussing, D.L.; Kolthoff, I.M. The solubility product of ferrous hydroxide and the ionization of the aquo-ferrous ion. J. Am. Chem. Soc. 1953, 75, 2476–2479. [Google Scholar] [CrossRef]
- CRC Handbook of Chemistry & Physics, 84th ed.; CRC Press: Boca Raton, FL, USA, 2003; p. 1314.
- Mellor, J.W. A Comprehensive Treatise on Inorganic and Theoretical Chemistry; Longmans, Green & Co.: London, UK, 1934; Volume XIII, pp. 929–937. [Google Scholar]
- Cheung, P.C.W.; Williams, D.R.; Kirk, D.W.; Barker, J. Minimization of Energy Wastage in the Electrochemical Recovery of Copper from Its EDTA Complexes in Wastewaters. Am. J. Environ. Eng. Sci. 2018, 5, 56–71. [Google Scholar]
- Gattrell, M.; Kirk, D. The electrochemical oxidation of aqueous phenol at a glassy carbon electrode. Can. J. Chem. Eng. 1990, 68, 997–1003. [Google Scholar] [CrossRef]
- Lamb, A.B.; Jacques, A.O. The slow hydrolysis of ferric chloride in dilute solution. II. The change in hydrogen ion concentration. Am. Chem. Soc. J. 1938, 60, 1215. [Google Scholar] [CrossRef]
- Hem, J.D.; Cropper, W.H. Survey of ferrous-ferric chemical equilibria and redox potentials. In Chemistry of Iron in Natural Water; Geological Survey Water Supply Paper 1459; U.S. Geological Survey: Washington, DC, USA, 1962; p. 9. [Google Scholar]
- Sarojam, P. Application Note: ICP Optical Emission Spectroscopy; Perkin-Elmer: Waltham, MA, USA, 2010; p. 6. Available online: https://www.perkinelmer.com/lab-solutions/resources/docs/APP_MetalsinWastewater.pdf (accessed on 1 July 2021).
- Stirling, G.C.; Ludman, C.J.; Waddington, T.C. Inelastic Neutron Scattering Spectra and Raman Spectra of CsHCl2 and CsDCl2. J. Chem. Phys. 1970, 52, 2730–2735. [Google Scholar] [CrossRef]
- Retgers, J.W. Beiträge zur Kenntnis des Isomorphismus VII: “XVI. Der Isomorphismus der Ferrate mit den Sulfaten, Seleniaten etc”. Z. Phys. Chem. 1892, 10, 529–557. [Google Scholar] [CrossRef]
- Horvath, R.; Keith, C.G. Understanding excited-state structure in metal polypyridyl complexes using resonance Raman excitation profiles, time-resolved resonance Raman spectroscopy and density functional theory. Coord. Chem. Rev. 2010, 254, 2505–2518. [Google Scholar] [CrossRef]
- Molchanova, A.D.; Prosnikov, M.A.; Petrov, V.P.; Dubrovin, R.M.; Nefedov, S.G.; Chernyshov, D.; Smirnov, A.N.; Davydov, V.Y.; Boldyrev, K.N.; Chernyshev, V.A.; et al. Lattice dynamics of cobalt orthoborate Co3(BO3)2 with kotoite structure. J. Alloy. Compd. 2021, 865, 158797. [Google Scholar] [CrossRef]
- Zając, A.; Dymińska, L.; Lorenc, J.; Kaczmarek, S.M.; Leniec, G.; Ptak, M.; Hanuza, J. Spectroscopic properties and molecular structure of copper phytate complexes: IR, Raman, UV–Vis, EPR studies and DFT calculations. J. Biol. Inorg. Chem. 2018, 24, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baiardi, A.; Latouche, C.; Bloino, J.; Barone, V. Accurate yet feasible computations of resonance Raman spectra for metal complexes in solution: [Ru(bpy)3]2+ as a case study. Dalton Trans. 2014, 43, 17610–17614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billes, F.; Holmgren, A.; Mikosch, H. A combined DFT and vibrational spectroscopy study of the nickel and zinc O,O-diethyldithiophosphate complexes. Vib. Spectrosc. 2010, 53, 296–306. [Google Scholar] [CrossRef]
- Pedesseau, L.; Chaix-Pluchery, O.; Modreanu, M.; Chaussende, D.; Sarigiannidou, E.; Rolland, A.; Even, J.; Durand, O. Al4SiC4 vibrational properties: Density functional theory calculations compared to Raman and infrared spectroscopy measurements. J. Raman Spectrosc. 2017, 48, 891–896. [Google Scholar] [CrossRef] [Green Version]
- Goodwill, J.E.; Jiang, Y.J.; Reckhow, D.A.; Gikonyo, J.; Tobiason, J.E. Characterization of Particles from Ferrate Preoxidation. Environ. Sci. Technol. 2015, 49, 4955–4962. [Google Scholar] [CrossRef]
- Amirbahman, A.; Sigg, L.; von Gunten, U. Reductive dissolution of Fe(III) hydroxides by cysteine: Kinetics and mechanism. J. Colloid Interface Sci. 1997, 194, 194–206. [Google Scholar] [CrossRef]
- Bzdyra, B.M.; Spellman, C.D.; Andreu, I.; Goodwill, J.E. Sulfite activation changes character of ferrate resultant particles. Chem. Eng. J. 2020, 393, 124771. [Google Scholar] [CrossRef]
- Barisci, S.; Dimoglo, A. Review on the Stability of Ferrate (VI) Species in Aqueous Medium and Oxidation of Pharmaceuticals and Personal Care Products (PPCPs) by Ferrate (VI): Identification of Transformation By-Products. In Ferrites and Ferrates: Chemistry and Applications in Sustainable Energy and Environmental Remediation; Chapter 12; American Chemical Society: Washington, DC, USA, 2016; Volume 1238, pp. 287–335. [Google Scholar] [CrossRef]
- Wang, S.; Shao, B.; Qiao, J.; Guan, X. Application of Fe(VI) in abating contaminants in water: State of art and knowledge gaps. Front. Environ. Sci. Eng. 2020, 15, 1–21. [Google Scholar]
- Hoffmann, R.; Alvarez, S.; Mealli, C.; Falceto, A.; Cahill, T.J.; Zeng, T.; Manca, G. From Widely Accepted Concepts in Coordination Chemistry to Inverted Ligand Fields. Chem. Rev. 2016, 116, 8173–8192. [Google Scholar] [CrossRef] [PubMed]
- Carsch, K.M.; Dimucci, I.M.; Iovan, D.A.; Li, A.; Zheng, S.L.; Titus, C.J.; Lee, S.J.; Irwin, K.D.; Nordlund, D.; Lancaster, K.M.; et al. Synthesis of a copper-supported triplet nitrene complex pertinent to copper−catalyzed amination. Science 2019, 365, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Bour, J.R.; Camasso, N.M.; Sanford, M.S. Oxidation of Ni(II) to Ni(IV) with Aryl Electrophiles Enables Ni-Mediated Aryl-CF3 Coupling. J. Am. Chem. Soc. 2015, 137, 8034–8037. [Google Scholar] [CrossRef] [PubMed]
- Dingle, A. Praseodymium unpaired. Nat. Chem. 2018, 10, 576. [Google Scholar] [CrossRef]
- Khun, T. Structure of Scientific Revolutions; 50th Anniversary Edition; University of Chicago Press: Chicago, IL, USA, 2012. [Google Scholar]
- Wolfsberg, M.W.; Helmholtz, L. The Spectra and Electronic Structure of the Tetrahedral Ions. J. Chem. Phys. 1952, 20, 837–843. [Google Scholar] [CrossRef]
- Ballhausen, C.A.; Gray, H.B. Molecular orbital theory: An introductory lecture note and reprint volume. In Frontiers in Chemistry; W. A. Benjamin, Inc.: New York, NY, USA, 1965. [Google Scholar]
- Schmidbaur, H.; Schwarz, E. Electronic Structure and Spectroscopic Properties of Group-7 Tri-Oxo-Halides. Inorg. Chem. 2021, 60, 9504–9515. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
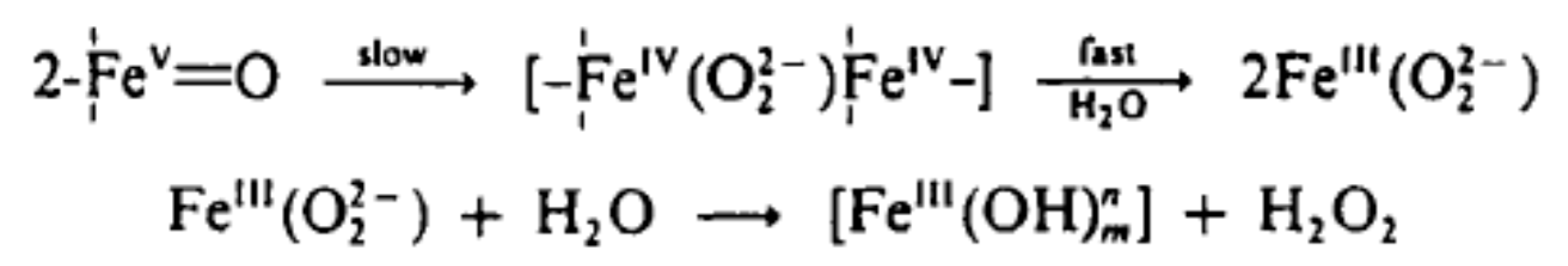{kind=link}
{kind=link}
{kind=link}
| Authors | Tarte/Nizet [84] | Griffith [44] | Gonzalez/Griffith [78] | Maghraoui et al. [86] |
|---|---|---|---|---|
| Year | 1963 | 1966 | 1972 | 2015 |
| Solid compound | K2FeO4 | K2FeO4 | K2FeO4 | Na2FeO4 |
| ν1 (Raman) | - | - | 830(10) | - |
| ν1 (i.r.) | 782 | 779 m (but disappears in D2O) | - | ~750 |
| ν2 (Raman) | - | - | 336(½) | - |
| ν2 (i.r.) | Possible association with ν4 (340, 322) | - | 340 m | (not reported) |
| ν3 (Raman) | - | - | 796(6) 786(1) | - |
| ν3 (i.r.) | 809 (singlet) 825 (small shoulder) | 827 sh 810 s 796 s (merges in D2O as one band at 800) s | 816 w 796 vs 780 m | 825 s (The 870 peak in [61] is actually from K2CrO4). |
| ν1 + ν3 (Raman) | - | - | - | - |
| ν1 + ν3 (i.r.) | (not reported) | 1570 w | (not reported) | (not reported) |
| ν4 (Raman) | - | - | 318(2) 312(3) 307(1) | - |
| ν4 (i.r.) | 340 w 322 w | 339 m 320 s (only the 320 peak remains in D2O) | 324 w 319 vs 311 w | (not reported) |
| ν2 + ν4 (Raman) | - | - | (not reported) | - |
| ν2 + ν4 (i.r.) | (see ν4 above) | - | (not reported) | (not reported) |
| Other strong bands (Raman) | - | (see two boxes to the right) | 840(2) | - |
| Other strong bands (i.r.) | (not reported) | (see two boxes to the right) | 620 s 297 w | 1140, 950, 930, 860, 620 (all suphate related). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheung, P.C.W.; Williams, D.R.; Barrett, J.; Barker, J.; Kirk, D.W. On the Origins of Some Spectroscopic Properties of “Purple Iron” (the Tetraoxoferrate(VI) Ion) and Its Pourbaix Safe-Space. Molecules 2021, 26, 5266. https://doi.org/10.3390/molecules26175266
Cheung PCW, Williams DR, Barrett J, Barker J, Kirk DW. On the Origins of Some Spectroscopic Properties of “Purple Iron” (the Tetraoxoferrate(VI) Ion) and Its Pourbaix Safe-Space. Molecules. 2021; 26(17):5266. https://doi.org/10.3390/molecules26175266
Chicago/Turabian StyleCheung, Philip C.W., Daryl R. Williams, Jack Barrett, James Barker, and Donald W. Kirk. 2021. "On the Origins of Some Spectroscopic Properties of “Purple Iron” (the Tetraoxoferrate(VI) Ion) and Its Pourbaix Safe-Space" Molecules 26, no. 17: 5266. https://doi.org/10.3390/molecules26175266
APA StyleCheung, P. C. W., Williams, D. R., Barrett, J., Barker, J., & Kirk, D. W. (2021). On the Origins of Some Spectroscopic Properties of “Purple Iron” (the Tetraoxoferrate(VI) Ion) and Its Pourbaix Safe-Space. Molecules, 26(17), 5266. https://doi.org/10.3390/molecules26175266