
CB1 Cannabinoid Receptor Signaling and Biased Signaling


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The CB1 Receptor
2.1. Therapeutic Potential
2.2. CB1 Physiology
2.3. Toxicity and Adverse Effects
3. CB1 Mechanism of Activation
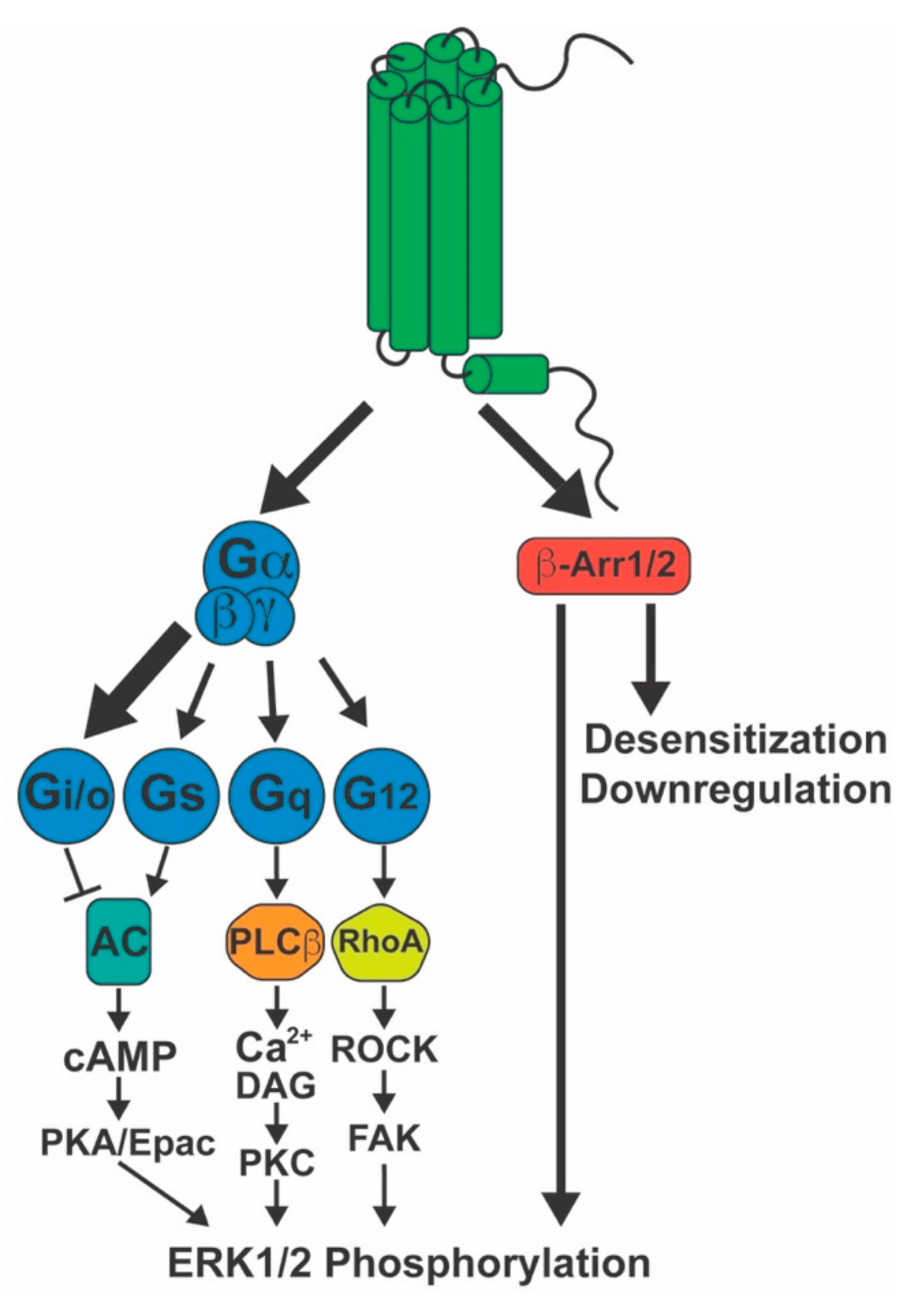
4. CB1 Signaling
4.1. G-Proteins
4.2. β-Arrestins
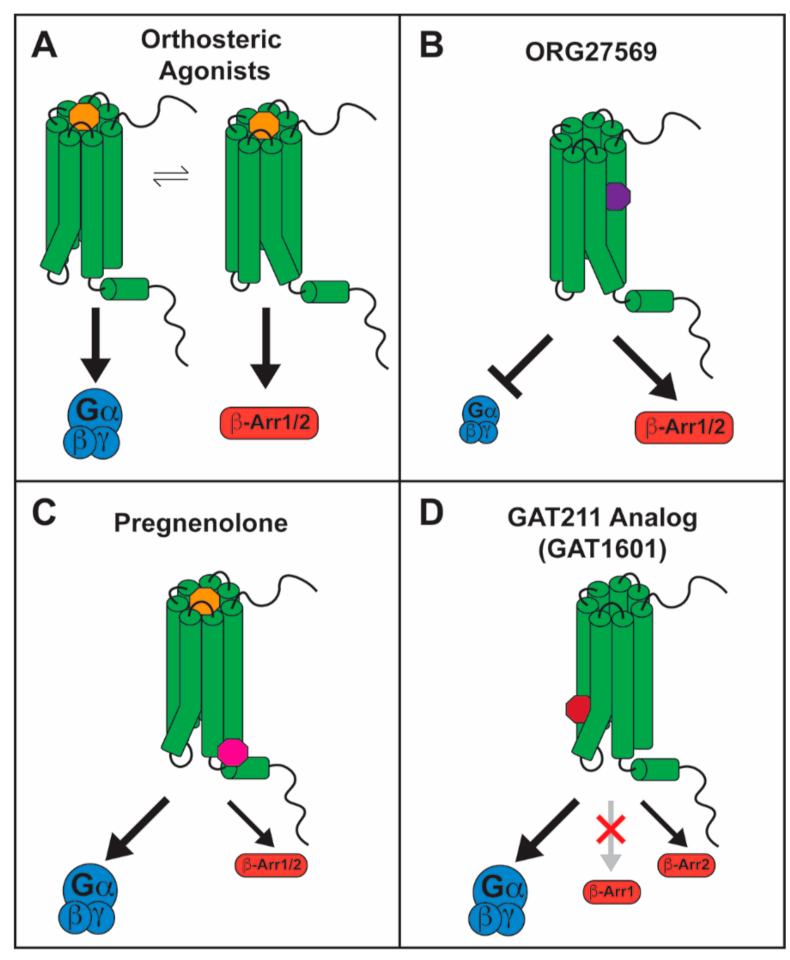
5. CB1-Biased Signaling
5.1. Orthosteric Ligands
5.2. Allosteric Ligands
5.2.1. ORG27569 as a Biased Allosteric Modulator of CB1
5.2.2. Pregnenolone as a Biased Allosteric Modulator of CB1
5.2.3. GAT211 as a Positive Allosteric Modulator of CB1
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and Characterization of a Canna-Binoid Receptor in Rat Brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nat. Cell Biol. 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Gérard, C.M.; Mollereau, C.; Vassart, G.; Parmentier, M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem. J. 1991, 279, 129–134. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Howlett, A.C. Pharmacology of Cannabinoid Receptors. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 607–634. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.-Q.; Melvin, L.S.; Makriyannis, A. The Conformational Properties of the Highly Selective Cannabinoid Receptor Ligand CP-55,940. J. Biol. Chem. 1996, 271, 10640–10647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissenstat, M.A.; Bell, M.R.; D’Ambra, T.E.; Alexander, E.J.; Daum, S.J.; Ackerman, J.H.; Gruett, M.D.; Kumar, V.; Estep, K.G. Aminoalkylindoles: Structure-Activity Relationships of Novel Cannabinoid Mimetics. J. Med. Chem. 1995, 38, 3094–3105. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylgylcerol: A Possible Endogenous Cannabinoid Receptor Ligand in Brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef]
- Smith, J.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Nurmikko, T.J.; Serpell, M.G.; Hoggart, B.; Toomey, P.J.; Morlion, B.J.; Haines, D. Sativex successfully treats neuropathic pain characterised by allodynia: A randomised, double-blind, placebo-controlled clinical trial. Pain 2007, 133, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Woodhams, S.G.; Chapman, V.; Finn, D.P.; Hohmann, A.G.; Neugebauer, V. The cannabinoid system and pain. Neuropharmacology 2017, 124, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, A.; Herkenham, M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: A double-label in situ hybridization study. Neuroscience 1999, 90, 923–931. [Google Scholar] [CrossRef]
- Ahluwalia, J.; Urban, L.; Capogna, M.; Bevan, S.J.; Nagy, I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience 2000, 100, 685–688. [Google Scholar] [CrossRef]
- Rahn, E.J.; Makriyannis, A.; Hohmann, A.G. Activation of cannabinoid CB1 and CB2 receptors suppresses neuropathic nociception evoked by the chemotherapeutic agent vincristine in rats. Br. J. Pharmacol. 2007, 152, 765–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernía-Andrade, A.J.; Kato, A.; Witschi, R.; Nyilas, R.; Katona, I.; Freund, T.F.; Watanabe, M.; Filitz, J.; Koppert, W.; Schüttler, J.; et al. Spinal Endocannabinoids and CB1 Receptors Mediate C-Fiber-Induced Heterosynaptic Pain Sensitization. Science 2009, 325, 760–764. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Xu, Q.; Shu, B.; Tiwari, V.; He, S.-Q.; Vera-Portocarrero, L.P.; Dong, X.; Linderoth, B.; Raja, S.N.; Wang, Y.; et al. Activation of cannabinoid CB1 receptor contributes to suppression of spinal nociceptive transmission and inhibition of mechanical hypersensitivity by Aβ-fiber stimulation. Pain 2016, 157, 2582–2593. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, A.H.; Cook, S.A.; Martin, B.R. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: Evidence supporting periaqueductal gray involvement. J. Pharmacol. Exp. Ther. 1996, 276, 585–593. [Google Scholar]
- Finn, D.; Jhaveri, M.; Beckett, S.; Roe, C.; Kendall, D.; Marsden, C.; Chapman, V. Effects of direct periaqueductal grey administration of a cannabinoid receptor agonist on nociceptive and aversive responses in rats. Neuropharmacology 2003, 45, 594–604. [Google Scholar] [CrossRef]
- Hohmann, A.G.; Suplita, R.L.; Bolton, N.M.; Neely, M.H.; Fegley, D.; Mangieri, R.; Krey, J.F.; Walker, J.M.; Holmes, P.V.; Crystal, J.D.; et al. An endocannabinoid mechanism for stress-induced analgesia. Nat. Cell Biol. 2005, 435, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Broyd, S.J.; Van Hell, H.H.; Beale, C.; Yücel, M.; Solowij, N. Acute and Chronic Effects of Cannabinoids on Human Cognition—A Systematic Review. Biol. Psychiatry 2016, 79, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Bilkei-Gorzo, A.; Albayram, O.; Draffehn, A.; Michel, K.; Piyanova, A.; Oppenheimer, H.; Dvir-Ginzberg, M.; Rácz, I.; Ulas, T.; Imbeault, S.; et al. A chronic low dose of Δ9-tetrahydrocannabinol (THC) restores cognitive function in old mice. Nat. Med. 2017, 23, 782–787. [Google Scholar] [CrossRef]
- Bilkei-Gorzo, A. The endocannabinoid system in normal and pathological brain ageing. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3326–3341. [Google Scholar] [CrossRef]
- Bilkei-Gorzo, A.; Racz, I.; Valverde, O.; Otto, M.; Michel, K.; Sarstre, M.; Zimmer, A. Early age-related cognitive impairment in mice lacking cannabinoid CB1 receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 15670–15675. [Google Scholar] [CrossRef] [Green Version]
- Haller, J.; Varga, B.; Ledent, C.; Freund, T.F. CB1 cannabinoid receptors mediate anxiolytic effects: Convergent genetic and pharmacological evidence with CB1-specific agents. Behav. Pharmacol. 2004, 15, 299–304. [Google Scholar] [CrossRef]
- Naderi, N.; Haghparast, A.; Saber-Tehrani, A.; Rezaii, N.; Alizadeh, A.-M.; Khani, A.; Motamedi, F. Interaction between cannabinoid compounds and diazepam on anxiety-like behaviour of mice. Pharmacol. Biochem. Behav. 2008, 89, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Carrier, E.J.; McLaughlin, R.; Morrish, A.C.; Meier, S.E.; Hillard, C.J.; Gorzalka, B.B. Regional alterations in the endocannabinoid system in an animal model of depression: Effects of concurrent antidepressant treatment. J. Neurochem. 2008, 106, 2322–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.-J.; Zheng, D.; Li, K.-X.; Yang, J.-M.; Pan, H.-Q.; Yu, X.-D.; Fu, J.-Y.; Zhu, Y.; Sun, Q.-X.; Tang, M.-Y.; et al. Cannabinoid CB1 receptors in the amygdalar cholecystokinin glutamatergic afferents to nucleus accumbens modulate depressive-like behavior. Nat. Med. 2019, 25, 337–349. [Google Scholar] [CrossRef]
- Sbarski, B.; Akirav, I. Cannabinoids as therapeutics for PTSD. Pharmacol. Ther. 2020, 211, 107551. [Google Scholar] [CrossRef]
- Wallace, M.; Wiley, J.; Martin, B.R.; DeLorenzo, R.J. Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur. J. Pharmacol. 2001, 428, 51–57. [Google Scholar] [CrossRef]
- Bahremand, A.; Nasrabady, S.E.; Shafaroodi, H.; Ghasemi, M.; Dehpour, A.R. Involvement of nitrergic system in the anticonvulsant effect of the cannabinoid CB1 agonist ACEA in the pentylenetetrazole-induced seizure in mice. Epilepsy Res. 2009, 84, 110–119. [Google Scholar] [CrossRef]
- Parmentier-Batteur, S.; Jin, K.; Mao, X.O.; Xie, L.; Greenberg, D.A. Increased Severity of Stroke in CB1 Cannabinoid Receptor Knock-Out Mice. J. Neurosci. 2002, 22, 9771–9775. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Mishima, K.; Nozako, M.; Hazekawa, M.; Ogata, A.; Fujioka, M.; Harada, K.; Mishima, S.; Orito, K.; Egashira, N.; et al. Δ9-tetrahydrocannabinol (Δ9-THC) prevents cerebral infarction via hypothalamic-independent hypothermia. Life Sci. 2007, 80, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jia, J.; Niu, W.; Jiang, T.; Zhai, Q.; Yang, L.; Bai, F.; Wang, Q.; Xiong, L. Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions. Sci. Rep. 2015, 5, 12440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, C.A.; Tabrizi, S. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Denovan-Wright, E.M.; Robertson, H.A. Cannabinoid receptor messenger RNA levels decrease in a subset of neurons of the lateral striatum, cortex and hippocampus of transgenic Huntington’s disease mice. Neuroscience 2000, 98, 705–713. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Berrendero, F.; Lucas, J.J.; Martín-Aparicio, E.; Yamamoto, A.; Ramos, J.; Fernández-Ruiz, J.J. Loss of mRNA levels, binding and activation of GTP-binding proteins for cannabinoid CB1 receptors in the basal ganglia of a transgenic model of Huntington’s disease. Brain Res. 2002, 929, 236–242. [Google Scholar] [CrossRef]
- Glass, M.; Faull, R.; Dragunow, M. Loss of cannabinoid receptors in the substantia nigra in huntington’s disease. Neuroscience 1993, 56, 523–527. [Google Scholar] [CrossRef]
- Chiarlone, A.; Bellocchio, L.; Blázquez, C.; Resel, E.; Soria-Gomez, E.; Cannich, A.; Ferrero, J.J.; Sagredo, O.; Benito, C.; Romero, J.; et al. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc. Natl. Acad. Sci. USA 2014, 111, 8257–8262. [Google Scholar] [CrossRef] [Green Version]
- Howlett, A. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nat. Cell Biol. 2001, 410, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Castillo, P.E.; Manzoni, O.J.; Tonini, R. Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 2017, 124, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Pacioni, S.; Bisogno, T.; Di Marzo, V.; Prince, D.A.; Huguenard, J.R.; Bacci, A. The Endocannabinoid 2-Arachidonoylglycerol Is Responsible for the Slow Self-Inhibition in Neocortical Interneurons. J. Neurosci. 2008, 28, 13532–13541. [Google Scholar] [CrossRef] [Green Version]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gomez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Han, J.; Kesner, P.; Metna-Laurent, M.; Duan, T.; Xu, L.; Georges, F.; Koehl, M.; Abrous, N.; Mendizabal-Zubiaga, J.; Grandes, P.; et al. Acute Cannabinoids Impair Working Memory through Astroglial CB1 Receptor Modulation of Hippocampal LTD. Cell 2012, 148, 1039–1050. [Google Scholar] [CrossRef] [Green Version]
- Metna-Laurent, M.; Marsicano, G. Rising stars: Modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia 2014, 63, 353–364. [Google Scholar] [CrossRef]
- Robin, L.M.; da Cruz, J.F.O.; Langlais, V.C.; Martin-Fernandez, M.; Metna-Laurent, M.; Busquets-Garcia, A.; Bellocchio, L.; Soria-Gomez, E.; Papouin, T.; Varilh, M.; et al. Astroglial CB1 Receptors Determine Synaptic D-Serine Availability to Enable Recognition Memory. Neuron 2018, 98, 935–944.e5. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Blasco, D.; Busquets-Garcia, A.; Hebert-Chatelain, E.; Serrat, R.; Vicente-Gutierrez, C.; Ioannidou, C.; Gómez-Sotres, P.; Lopez-Fabuel, I.; Resch-Beusher, M.; Resel, E.; et al. Glucose metabolism links astroglial mitochondria to cannabinoid effects. Nat. Cell Biol. 2020, 583, 603–608. [Google Scholar] [CrossRef]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K. Distribution of Cannabinoid Receptors in the Central and Peripheral Nervous System. In Cannabinoids. Handbook of Experimental Pharmacology; Pertwee, R.G., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 168, pp. 299–325. [Google Scholar]
- Kelly, B.F.; Nappe, T.M. Cannabinoid Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- O’Sullivan, S.E. Endocannabinoids and the Cardiovascular System in Health and Disease. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 231, pp. 393–422. [Google Scholar]
- Tang, X.; Liu, Z.; Li, X.; Wang, J.; Li, L. Cannabinoid Receptors in Myocardial Injury: A Brother Born to Rival. Int. J. Mol. Sci. 2021, 22, 6886. [Google Scholar] [CrossRef]
- Koch, M.; Varela, L.; Kim, J.G.; Kim, J.D.; Hernández-Nuño, F.; Simonds, S.; Castorena, C.M.; Vianna, C.R.; Elmquist, J.K.; Morozov, Y.; et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nat. Cell Biol. 2015, 519, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Cota, D.; Marsicano, G.; Tschöp, M.; Grübler, Y.; Flachskamm, C.; Schubert, M.; Auer, D.; Yassouridis, A.; Thöne-Reineke, C.; Ortmann, S.; et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Investig. 2003, 112, 423–431. [Google Scholar] [CrossRef]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bátkai, S.; Harvey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Zhang, G.; Mou, C.; Fu, X.; Chen, Y. Peripheral CB1 Receptor Neutral Antagonist, AM6545, Ameliorates Hypometabolic Obesity and Improves Adipokine Secretion in Monosodium Glutamate Induced Obese Mice. Front. Pharmacol. 2018, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Paszkiewicz, R.L.; Bergman, R.N.; Santos, R.S.; Frank, A.P.; Woolcott, O.O.; Iyer, M.S.; Stefanovski, D.; Clegg, D.J.; Kabir, M. A Peripheral CB1R Antagonist Increases Lipolysis, Oxygen Consumption Rate, and Markers of Beiging in 3T3-L1 Adipocytes Similar to RIM, Suggesting that Central Effects Can Be Avoided. Int. J. Mol. Sci. 2020, 21, 6639. [Google Scholar] [CrossRef]
- Müller, G.A.; Herling, A.W.; Wied, S.; Müller, T.D. CB1 Receptor-Dependent and Independent Induction of Lipolysis in Primary Rat Adipocytes by the Inverse Agonist Rimonabant (SR141716A). Molecules 2020, 25, 896. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.-S.; et al. Peripheral Cannabinoid-1 Receptor Inverse Agonism Reduces Obesity by Reversing Leptin Resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.; Szanda, G.; Drori, A.; Liu, Z.; Cinar, R.; Kashiwaya, Y.; Reitman, M.L.; Kunos, G. Peripheral cannabinoid-1 receptor blockade restores hypothalamic leptin signaling. Mol. Metab. 2017, 6, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Drori, A.; Gammal, A.; Azar, S.; Hinden, L.; Hadar, R.; Wesley, D.; Nemirovski, A.; Szanda, G.; Salton, M.; Tirosh, B.; et al. CB1R regulates soluble leptin receptor levels via CHOP, contributing to hepatic leptin resistance. eLife 2020, 9, 60771. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, U.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.-I.; Bátkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Azar, S.; Udi, S.; Drori, A.; Hadar, R.; Nemirovski, A.; Vemuri, K.V.; Miller, M.; Sherill-Rofe, D.; Arad, Y.; Gur-Wahnon, D.; et al. Reversal of diet-induced hepatic steatosis by peripheral CB1 receptor blockade in mice is p53/miRNA-22/SIRT1/PPARα dependent. Mol. Metab. 2020, 42, 101087. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Laudermilk, L.; Ware, J.; Rosa, T.; Mathews, K.; Gay, E.; Amato, G.; Maitra, R. Peripherally Selective CB1 Receptor Antagonist Improves Symptoms of Metabolic Syndrome in Mice. ACS Pharmacol. Transl. Sci. 2021, 4, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lao, Q.; Shin, Y.-K.; Carlson, O.D.; Lee, E.K.; Gorospe, M.; Kulkarni, R.N.; Egan, J.M. Cannabinoids Induce Pancreatic -Cell Death by Directly Inhibiting Insulin Receptor Activation. Sci. Signal. 2012, 5, ra23. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, T.; Nicoloro, S.M.; Zhou, Z.; Shen, Y.; Liu, J.; Coffey, N.J.; Cinar, R.; Godlewski, G.; Gao, B.; Aouadi, M.; et al. Decreasing CB 1 receptor signaling in Kupffer cells improves insulin sensitivity in obese mice. Mol. Metab. 2017, 6, 1517–1528. [Google Scholar] [CrossRef]
- Eid, B.; Neamatallah, T.; Hanafy, A.; El-Bassossy, H.; Aldawsari, H.; Vemuri, K.; Makriyannis, A. Effects of the CB1 Receptor Antagonists AM6545 and AM4113 on Insulin Resistance in a High-Fructose High-Salt Rat Model of Metabolic Syndrome. Medicina 2020, 56, 573. [Google Scholar] [CrossRef]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Topol, E.J.; Bousser, M.-G.; Fox, K.; Creager, M.A.; Despres, J.-P.; Easton, J.D.; Hamm, C.W.; Montalescot, G.; Steg, P.G.; Pearson, T.A.; et al. Rimonabant for prevention of cardiovascular events (CRESCENDO): A randomised, multicentre, placebo-controlled trial. Lancet 2010, 376, 517–523. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nat. Cell Biol. 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Vemuri, K.; Nikas, S.P.; LaPrairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nat. Cell Biol. 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Kumar, K.K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Kapur, A.; Hurst, D.P.; Fleischer, D.; Whitnell, R.; Thakur, G.A.; Makriyannis, A.; Reggio, P.H.; Abood, M.E. Mutation Studies of Ser7.39 and Ser2.60 in the Human CB1 Cannabinoid Receptor: Evidence for a Serine-Induced Bend in CB1 Transmem-brane Helix 7. Mol. Pharm. 2007, 71, 1512–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, S.D.; Hurst, D.P.; Barnett-Norris, J.; Lynch, D.; Reggio, P.H.; Abood, M. Structural Mimicry in Class A G Protein-coupled Receptor Rotamer Toggle Switches. J. Biol. Chem. 2004, 279, 48024–48037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Antona, A.M.; Ahn, K.H.; Kendall, D.A. Mutations of CB1 T210 Produce Active and Inactive Receptor Forms: Correlations with Ligand Affinity, Receptor Stability, and Cellular Localization. Biochemistry 2006, 45, 5606–5617. [Google Scholar] [CrossRef] [Green Version]
- Cahill, T.J.; Thomsen, A.; Tarrasch, J.T.; Plouffe, B.; Nguyen, A.; Yang, F.; Huang, L.-Y.; Kahsai, A.W.; Bassoni, D.L.; Gavino, B.J.; et al. Distinct conformations of GPCR–β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2562–2567. [Google Scholar] [CrossRef] [Green Version]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nat. Cell Biol. 2020, 579, 297–302. [Google Scholar] [CrossRef]
- Gyombolai, P.; Tóth, A.D.; Tímár, D.; Turu, G.; Hunyady, L. Mutations in the ‘DRY’ motif of the CB1 cannabinoid receptor result in biased receptor variants. J. Mol. Endocrinol. 2014, 54, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased Signaling Pathways in 2-Adrenergic Receptor Characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [Green Version]
- Rahmeh, R.; Damian, M.; Cottet, M.; Orcel, H.; Mendre, C.; Durroux, T.; Sharma, K.S.; Durand, G.; Pucci, B.; Trinquet, E.; et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 6733–6738. [Google Scholar] [CrossRef] [Green Version]
- Fay, J.F.; Farrens, D.L. Structural dynamics and energetics underlying allosteric inactivation of the cannabinoid receptor CB1. Proc. Natl. Acad. Sci. USA 2015, 112, 8469–8474. [Google Scholar] [CrossRef] [Green Version]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.-P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nat. Cell Biol. 2015, 523, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, N.; Itoh, H. Functions and Regulatory Mechanisms of Gq-Signaling Pathways. Neurosignals 2009, 17, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 2009, 158, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldeeb, K.; Leone-Kabler, S.; Howlett, A.C. CB1 cannabinoid receptor-mediated increases in cyclic AMP accumulation are correlated with reduced Gi/o function. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 311–322. [Google Scholar] [CrossRef]
- Bonhaus, D.W.; Chang, L.K.; Kwan, J.; Martin, G.R. Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: Evidence for agonist-specific trafficking of intracellular responses. J. Pharmacol. Exp. Ther. 1998, 287, 884–888. [Google Scholar]
- Calandra, B.; Portier, M.; Kernéis, A.; Delpech, M.; Carillon, C.; Le Fur, G.; Ferrara, P.; Shire, D. Dual intracellular signaling pathways mediated by the human cannabinoid CB1 receptor. Eur. J. Pharmacol. 1999, 374, 445–455. [Google Scholar] [CrossRef]
- Glass, M.; Felder, C.C. Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Augments cAMP Accumulation in Striatal Neurons: Evidence for a GsLinkage to the CB1 Receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef] [Green Version]
- Lauckner, J.E.; Hille, B.; Mackie, K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19144–19149. [Google Scholar] [CrossRef] [Green Version]
- Ishii, I.; Chun, J. Anandamide-induced neuroblastoma cell rounding via the CB1 cannabinoid receptors. NeuroReport 2002, 13, 593–596. [Google Scholar] [CrossRef]
- Eldeeb, K.; Leone-Kabler, S.; Howlett, A.C. Mouse Neuroblastoma CB1 Cannabinoid Receptor-Stimulated [35S]GTPɣS Binding: Total and Antibody-Targeted Gα Protein-Specific Scintillation Proximity Assays. In Methods in Enzymology; Reggio, P.H., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 593, pp. 1–21. [Google Scholar]
- Roland, A.; Ricobaraza, A.; Carrel, D.; Jordan, B.M.; Rico, F.; Simon, A.; Humbert-Claude, M.; Ferrier, J.; McFadden, M.H.; Scheuring, S.; et al. Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. eLife 2014, 3, e03159. [Google Scholar] [CrossRef]
- Ibsen, M.S.; Finlay, D.; Patel, M.; Javitch, J.A.; Glass, M.; Grimsey, N.L. Cannabinoid CB1 and CB2 Receptor-Mediated Arrestin Translocation: Species, Subtype, and Agonist-Dependence. Front. Pharmacol. 2019, 10, 350. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Dupre, D.J.; Denovan-Wright, E.M. Type 1 Cannabinoid Receptor Ligands Display Functional Selectivity in a Cell Culture Model of Striatal Medium Spiny Projection Neurons. J. Biol. Chem. 2014, 289, 24845–24862. [Google Scholar] [CrossRef] [Green Version]
- Breivogel, C.; Childers, S.R.; Deadwyler, S.A.; Hampson, R.E.; Vogt, L.J.; Sim-Selley, L.J. Chronic delta9-Tetrahydrocannabinol Treatment Produces a Time-Dependent Loss of Cannabinoid Receptors and Cannabinoid Receptor-Activated G Proteins in Rat Brain. J. Neurochem. 2002, 73, 2447–2459. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.T.; Schmid, C.L.; Raehal, K.M.; Selley, D.E.; Bohn, L.M.; Sim-Selley, L.J. β-Arrestin2 Regulates Cannabinoid CB1 Receptor Signaling and Adaptation in a Central Nervous System Region–Dependent Manner. Biol. Psychiatry 2012, 71, 714–724. [Google Scholar] [CrossRef] [Green Version]
- Breivogel, C.S.; Puri, V.; Lambert, J.M.; Hill, D.K.; Huffman, J.W.; Razdan, R.K. The influence of beta-arrestin2 on cannabinoid CB1receptor coupling to G-proteins and subcellular localization and relative levels of beta-arrestin1 and 2 in mouse brain. J. Recept. Signal Transduct. 2013, 33, 367–379. [Google Scholar] [CrossRef]
- Breivogel, C.S.; Vaghela, M.S. The effects of beta-arrestin1 deletion on acute cannabinoid activity, brain cannabinoid receptors and tolerance to cannabinoids in mice. J. Recept. Signal Transduct. 2014, 35, 98–106. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. Transduction of Receptor Signals by -Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Shenoy, S.K.; Wei, H.; Lefkowitz, R.J. Differential Kinetic and Spatial Patterns of β-Arrestin and G Protein-mediated ERK Activation by the Angiotensin II Receptor. J. Biol. Chem. 2004, 279, 35518–35525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by -arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [Green Version]
- Tohgo, A.; Pierce, K.L.; Choy, E.W.; Lefkowitz, R.J.; Luttrell, L. β-Arrestin Scaffolding of the ERK Cascade Enhances Cytosolic ERK Activity but Inhibits ERK-mediated Transcription following Angiotensin AT1a Receptor Stimulation. J. Biol. Chem. 2002, 277, 9429–9436. [Google Scholar] [CrossRef] [Green Version]
- Caunt, C.J.; Finch, A.R.; Sedgley, K.R.; McArdle, C.A. Seven-transmembrane receptor signalling and ERK compartmentalization. Trends Endocrinol. Metab. 2006, 17, 276–283. [Google Scholar] [CrossRef]
- Metna-Laurent, M.; Mondésir, M.; Grel, A.; Vallée, M.; Piazza, P. Cannabinoid-Induced Tetrad in Mice. Curr. Protoc. Neurosci. 2017, 80, 9–59. [Google Scholar] [CrossRef]
- Breivogel, C.S.; Lambert, J.M.; Gerfin, S.; Huffman, J.W.; Razdan, R.K. Sensitivity to Δ9-tetrahydrocannabinol is selectively enhanced in beta-arrestin2−/− mice. Behav. Pharmacol. 2008, 19, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.; Wickens, A. Enhancement by chlordiazepoxide of catalepsy induced in rats by intravenous or intrapallidal injections of enantiomeric cannabinoids. Neuropharmacology 1991, 30, 237–244. [Google Scholar] [CrossRef]
- Wallmichrath, I.; Szabo, B. Cannabinoids inhibit striatonigral GABAergic neurotransmission in the mouse. Neuroscience 2002, 113, 671–682. [Google Scholar] [CrossRef]
- Ahn, K.H.; Mahmoud, M.; Shim, J.-Y.; Kendall, D.A. Distinct Roles of β-Arrestin 1 and β-Arrestin 2 in ORG27569-induced Biased Signaling and Internalization of the Cannabinoid Receptor 1 (CB1). J. Biol. Chem. 2013, 288, 9790–9800. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Brown, S.; Roche, J.P.; Hsieh, C.; Celver, J.P.; Kovoor, A.; Chavkin, C.; Mackie, K. Distinct Domains of the CB1 Cannabinoid Receptor Mediate Desensitization and Internalization. J. Neurosci. 1999, 19, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Daigle, T.L.; Kearn, C.S.; Mackie, K. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology 2008, 54, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Peraza, F.; Ahn, K.H.; Nogueras-Ortiz, C.; Mungrue, I.; Mackie, K.; Kendall, D.A.; Yudowski, G.A. Mechanisms of Biased β-Arrestin-Mediated Signaling Downstream from the Cannabinoid 1 Receptor. Mol. Pharmacol. 2016, 89, 618–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPrairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Biased Type 1 Cannabinoid Receptor Signaling Influences Neuronal Viability in a Cell Culture Model of Huntington Disease. Mol. Pharmacol. 2015, 89, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T.; Watson, C.; Muniz-Medina, V.; Christopoulos, A.; Novick, S. A Simple Method for Quantifying Functional Selectivity and Agonist Bias. ACS Chem. Neurosci. 2012, 3, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Khajehali, E.; Malone, D.T.; Glass, M.; Sexton, P.; Christopoulos, A.; Leach, K. Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol. Pharmacol. 2015, 88, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Finlay, D.B.; Glass, M.; Duffull, S.B. Evaluation of the profiles of CB 1 cannabinoid receptor signalling bias using joint kinetic modelling. Br. J. Pharmacol. 2020, 177, 3449–3463. [Google Scholar] [CrossRef]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem. 2019, 62, 88–127. [Google Scholar] [CrossRef]
- Price, M.R.; Baillie, G.L.; Thomas, A.; Stevenson, L.A.; Easson, M.; Goodwin, R.; McLean, A.; McIntosh, L.; Goodwin, G.; Walker, G.; et al. Allosteric Modulation of the Cannabinoid CB1 Receptor. Mol. Pharmacol. 2005, 68, 1484–1495. [Google Scholar] [CrossRef] [Green Version]
- Baillie, G.L.; Horswill, J.G.; Anavi-Goffer, S.; Reggio, P.H.; Bolognini, D.; Abood, M.; McAllister, S.D.; Strange, P.G.; Stephens, G.J.; Pertwee, R.; et al. CB1 Receptor Allosteric Modulators Display Both Agonist and Signaling Pathway Specificity. Mol. Pharmacol. 2013, 83, 322–338. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.H.; Mahmoud, M.M.; Kendall, D.A. Allosteric Modulator ORG27569 Induces CB1 Cannabinoid Receptor High Affinity Agonist Binding State, Receptor Internalization, and Gi Protein-Independent ERK1/2 Kinase Activation. J. Bioloical Chem. 2012, 287, 12070–12082. [Google Scholar] [CrossRef] [Green Version]
- Cawston, E.E.; Redmond, W.J.; Breen, C.M.; Grimsey, N.; Connor, M.; Glass, M. Real-time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br. J. Pharmacol. 2013, 170, 893–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straiker, A.; Mitjavila, J.; Yin, D.; Gibson, A.; Mackie, K. Aiming for allosterism: Evaluation of allosteric modulators of CB1 in a neuronal model. Pharmacol. Res. 2015, 99, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamage, T.F.; Anderson, J.C.; Abood, M.E. CB1 Allosteric Modulator Org27569 Is an Antagonist/Inverse Agonist of ERK1/2 Signaling. Cannabis Cannabinoid Res. 2016, 1, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef]
- Lynch, D.L.; Hurst, W.P.; Shore, D.M.; Pitman, M.C.; Reggio, P.H. Molecular Dynamics Methodologies for Probing Cannabinoid Ligand/Receptor Interaction. Methods Enzym. 2017, 593, 449–490. [Google Scholar] [CrossRef] [Green Version]
- Gamage, T.F.; Ignatowska-Jankowska, B.; Wiley, J.; Abdelrahman, M.; Trembleau, L.; Greig, I.; Thakur, G.A.; Tichkule, R.; Poklis, J.; Ross, R.A.; et al. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behav. Pharmacol. 2014, 25, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Qiu, Y.; Jing, L.; Thorn, D.A.; Zhang, Y.; Li, J.-X. Behavioral effects of the cannabinoid CB1receptor allosteric modulator ORG27569 in rats. Pharmacol. Res. Perspect. 2014, 2, e00069. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, A.; McMahon, L.R. In vivo pharmacology of endocannabinoids and their metabolic inhibitors: Therapeutic implications in Parkinson’s disease and abuse liability. Prostaglandins Other Lipid Mediat. 2010, 91, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Schulze, D.R.; McMahon, L.R. Tolerance and cross-tolerance to cannabinoids in mice: Schedule-controlled responding and hypothermia. Psychopharmacology 2011, 215, 665–675. [Google Scholar] [CrossRef] [Green Version]
- Vallee, M.; Vitiello, S.; Bellocchio, L.; Hébert-Chatelain, E.; Monlezun, S.; Martín-García, E.; Kasanetz, F.; Baillie, G.L.; Panin, F.; Cathala, A.; et al. Pregnenolone Can Protect the Brain from Cannabis Intoxication. Science 2014, 343, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Busquets-Garcia, A.; Soria-Gomez, E.; Redon, B.; Mackenbach, Y.; Vallee, M.; Chaouloff, F.; Varilh, M.; Ferreira, G.; Piazza, P.-V.; Marsicano, G. Pregnenolone blocks cannabinoid-induced acute psychotic-like states in mice. Mol. Psychiatry 2017, 22, 1594–1603. [Google Scholar] [CrossRef] [Green Version]
- Laprairie, R.B.; Kulkarni, P.M.; Deschamps, J.R.; Kelly, M.E.M.; Janero, D.R.; Cascio, M.G.; Stevenson, L.A.; Pertwee, R.G.; Kenakin, T.P.; Denovan-Wright, E.M.; et al. Enantiospecific Allosteric Modulation of Cannabinoid 1 Receptor. ACS Chem. Neurosci. 2017, 8, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Garai, S.; Kulkarni, P.M.; Schaffer, P.C.; Leo, L.M.; Brandt, A.L.; Zagzoog, A.; Black, T.; Lin, X.; Hurst, D.P.; Janero, D.R.; et al. Application of Fluorine- and Nitrogen-Walk Approaches: Defining the Structural and Functional Diversity of 2-Phenylindole Class of Cannabinoid 1 Receptor Positive Allosteric Modulators. J. Med. Chem. 2020, 63, 542–568. [Google Scholar] [CrossRef] [PubMed]
- Mitjavila, J.; Yin, D.; Kulkarni, P.M.; Zanato, C.; Thakur, G.A.; Ross, R.; Greig, I.; Mackie, K.; Straiker, A. Enantiomer-specific positive allosteric modulation of CB1 signaling in autaptic hippocampal neurons. Pharmacol. Res. 2018, 129, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, D.P.; Garai, S.; Kulkarni, P.M.; Schaffer, P.C.; Reggio, P.H.; Thakur, G.A. Identification of CB1 Receptor Allosteric Sites Using Force-Biased MMC Simulated Annealing and Validation by Structure–Activity Relationship Studies. ACS Med. Chem. Lett. 2019, 10, 1216–1221. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Rourke, J.L.; Zrein, A.; Cairns, E.A.; Kelly, M.E.; Sinal, C.J.; Kulkarni, P.M.; Thakur, G.A.; Denovan-Wright, E.M. Positive allosteric modulation of the type 1 cannabinoid receptor reduces the signs and symptoms of Huntington’s disease in the R6/2 mouse model. Neuropharmacology 2019, 151, 1–12. [Google Scholar] [CrossRef]
- Cairns, E.A.; Szczesniak, A.-M.; Straiker, A.J.; Kulkarni, P.M.; Pertwee, R.G.; Thakur, G.A.; Baldridge, W.H.; Kelly, M.E. The In Vivo Effects of the CB1-Positive Allosteric Modulator GAT229 on Intraocular Pressure in Ocular Normotensive and Hypertensive Mice. J. Ocul. Pharmacol. Ther. 2017, 33, 582–590. [Google Scholar] [CrossRef]
- Roebuck, A.J.; Greba, Q.; Smolyakova, A.-M.; Alaverdashvili, M.; Marks, W.N.; Garai, S.; Baglot, S.L.; Petrie, G.; Cain, S.M.; Snutch, T.P.; et al. Positive allosteric modulation of type 1 cannabinoid receptors reduces spike-and-wave discharges in Genetic Absence Epilepsy Rats from Strasbourg. Neuropharmacology 2021, 190, 108553. [Google Scholar] [CrossRef]
- Slivicki, R.A.; Xu, Z.; Kulkarni, P.M.; Pertwee, R.G.; Mackie, K.; Thakur, G.A.; Hohmann, A.G. Positive Allosteric Modulation of Cannabinoid Receptor Type 1 Suppresses Pathological Pain Without Producing Tolerance or Dependence. Biol. Psychiatry 2018, 84, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Slivicki, R.A.; Iyer, V.; Mali, S.S.; Garai, S.; Thakur, G.A.; Crystal, J.D.; Hohmann, A.G. Positive Allosteric Modulation of CB1 Cannabinoid Receptor Signaling Enhances Morphine Antinociception and Attenuates Morphine Tolerance Without Enhancing Morphine- Induced Dependence or Reward. Front. Mol. Neurosci. 2020, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Garai, S.; Leo, L.M.; Szczesniak, A.-M.; Hurst, D.P.; Schaffer, P.C.; Zagzoog, A.; Black, T.; Deschamps, J.R.; Miess, E.; Schulz, S.; et al. Discovery of a Biased Allosteric Modulator for Cannabinoid 1 Receptor: Preclinical Anti-Glaucoma Efficacy. J. Med. Chem. 2021, 64, 8104–8126. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Jensen, A.D.; Liapakis, G.; Rasmussen, S.; Shi, L.; Gether, U.; Javitch, J. Activation of the β2-Adrenergic Receptor Involves Disruption of an Ionic Lock between the Cytoplasmic Ends of Transmembrane Segments 3 and 6. J. Biol. Chem. 2001, 276, 29171–29177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.; Choi, H.-J.; DeVree, B.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nat. Cell Biol. 2011, 469, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.W.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nat. Cell Biol. 2016, 536, 104–107. [Google Scholar] [CrossRef]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science 2020, 367, 881–887. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leo, L.M.; Abood, M.E. CB1 Cannabinoid Receptor Signaling and Biased Signaling. Molecules 2021, 26, 5413. https://doi.org/10.3390/molecules26175413
Leo LM, Abood ME. CB1 Cannabinoid Receptor Signaling and Biased Signaling. Molecules. 2021; 26(17):5413. https://doi.org/10.3390/molecules26175413
Chicago/Turabian StyleLeo, Luciana M., and Mary E. Abood. 2021. "CB1 Cannabinoid Receptor Signaling and Biased Signaling" Molecules 26, no. 17: 5413. https://doi.org/10.3390/molecules26175413
APA StyleLeo, L. M., & Abood, M. E. (2021). CB1 Cannabinoid Receptor Signaling and Biased Signaling. Molecules, 26(17), 5413. https://doi.org/10.3390/molecules26175413