
Central Composite Design for Formulation and Optimization of Solid Lipid Nanoparticles to Enhance Oral Bioavailability of Acyclovir

 , ,
, ,
Abstract
:1. Introduction
2. Results
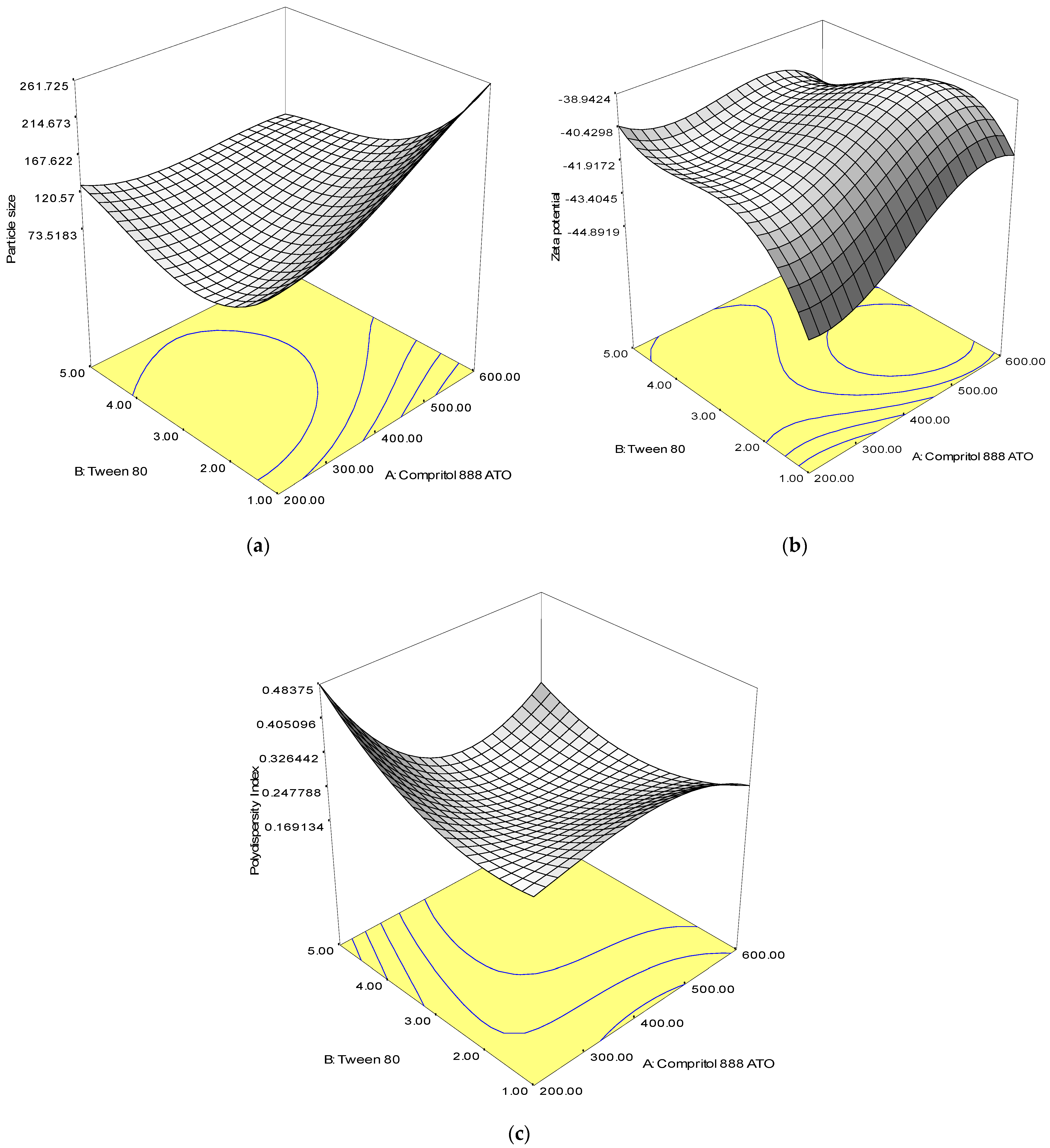
2.1. Fitting the Response Surface Model
2.2. Verification of Reduced Model
2.3. Physical Characteristics and Entrapment Efficiency of Acyclovir-Loaded SLN
2.4. Transmission Electron Microscopy
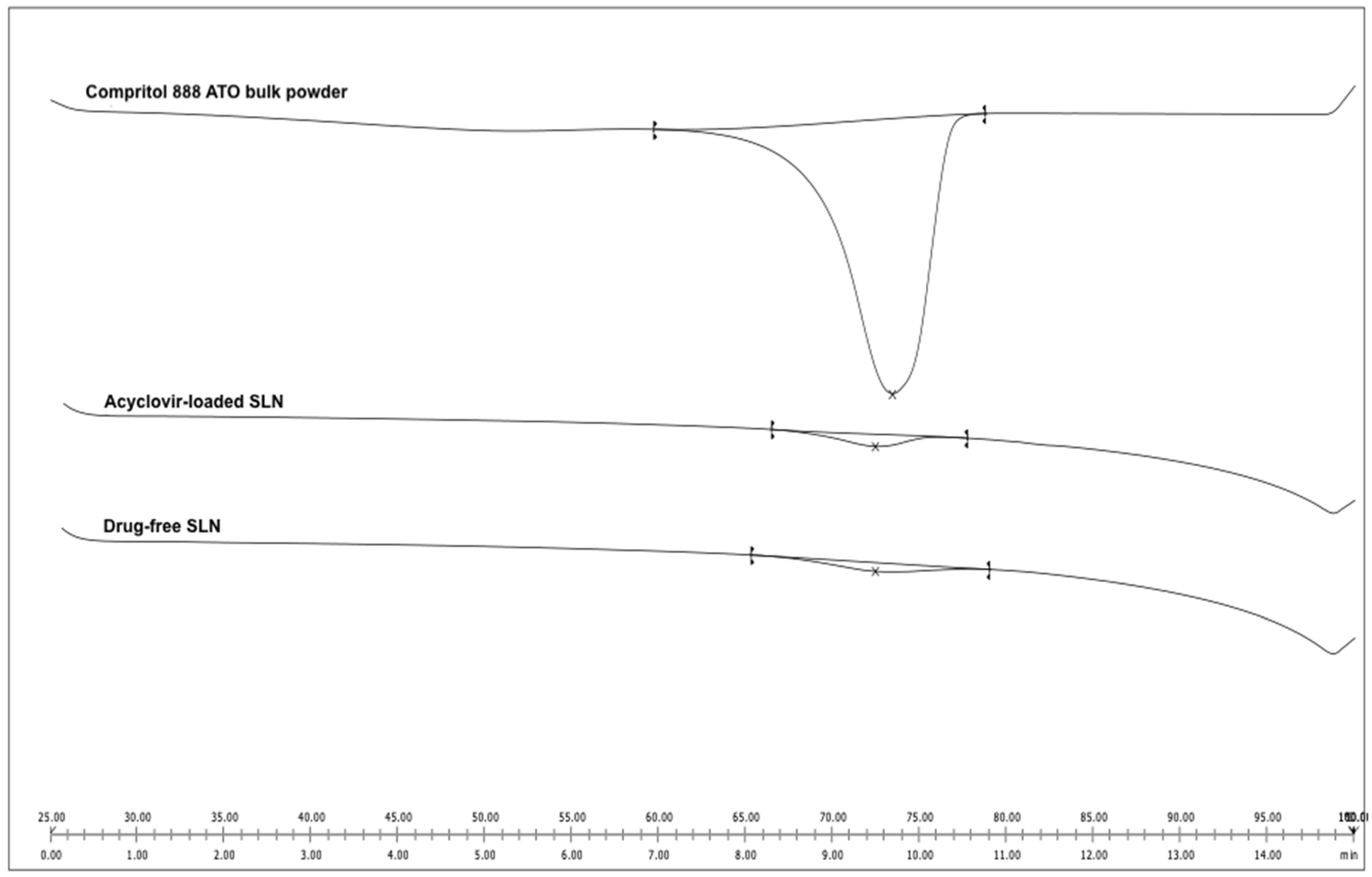
2.5. Differential Scanning Calorimetry Analysis
2.6. Short-Term Stability Test
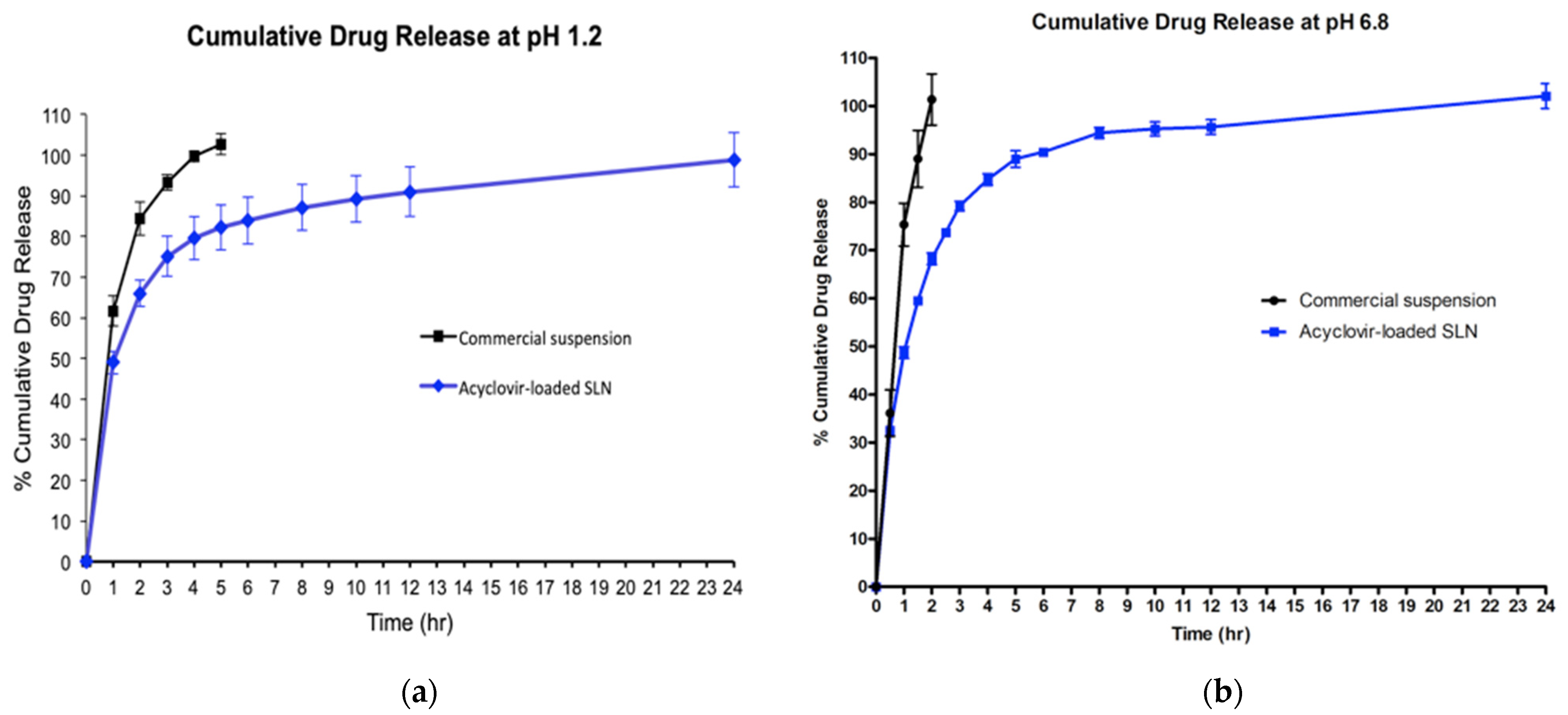
2.7. In Vitro Release Study
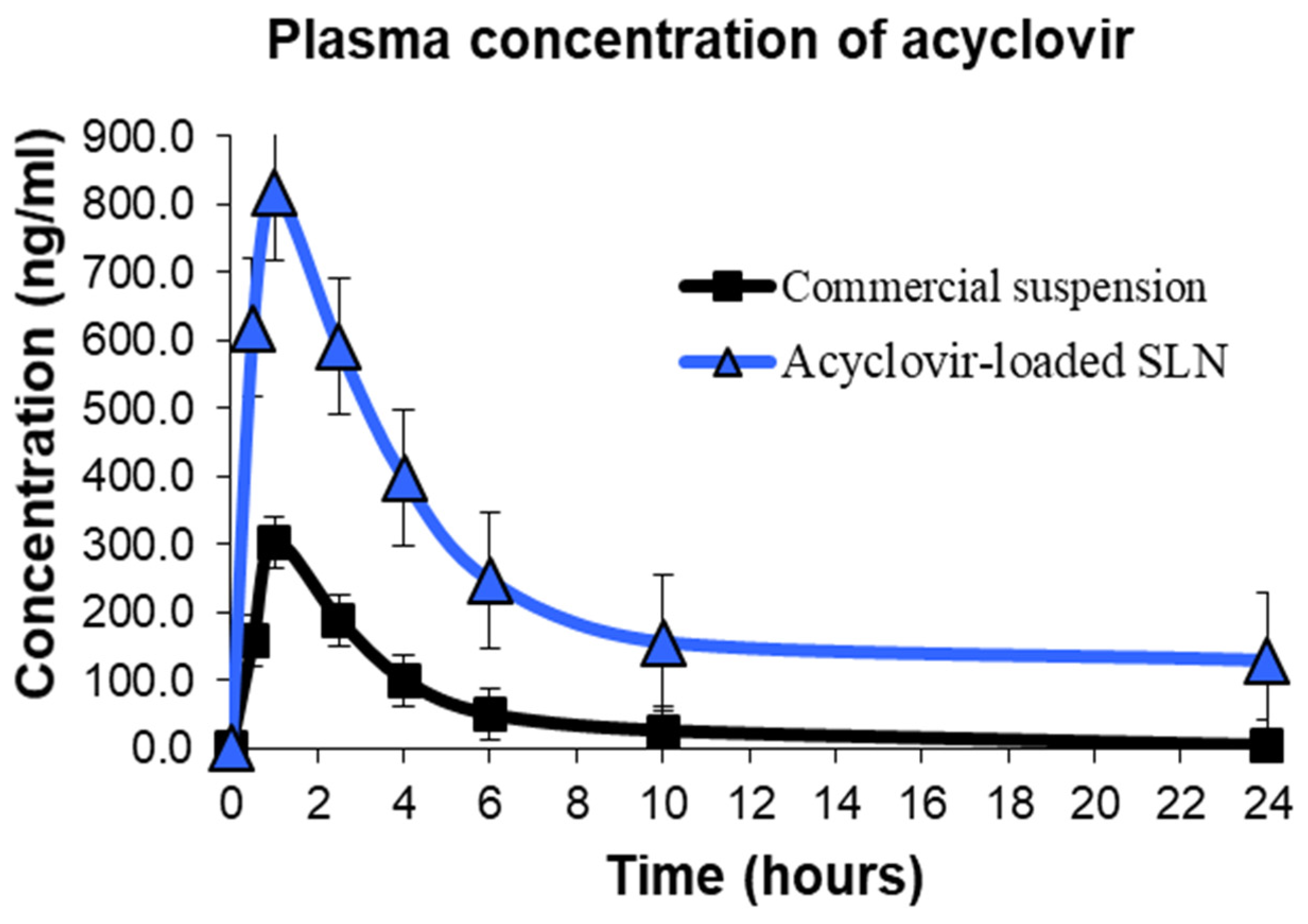
2.8. In Vivo Pharmacokinetic Evaluation
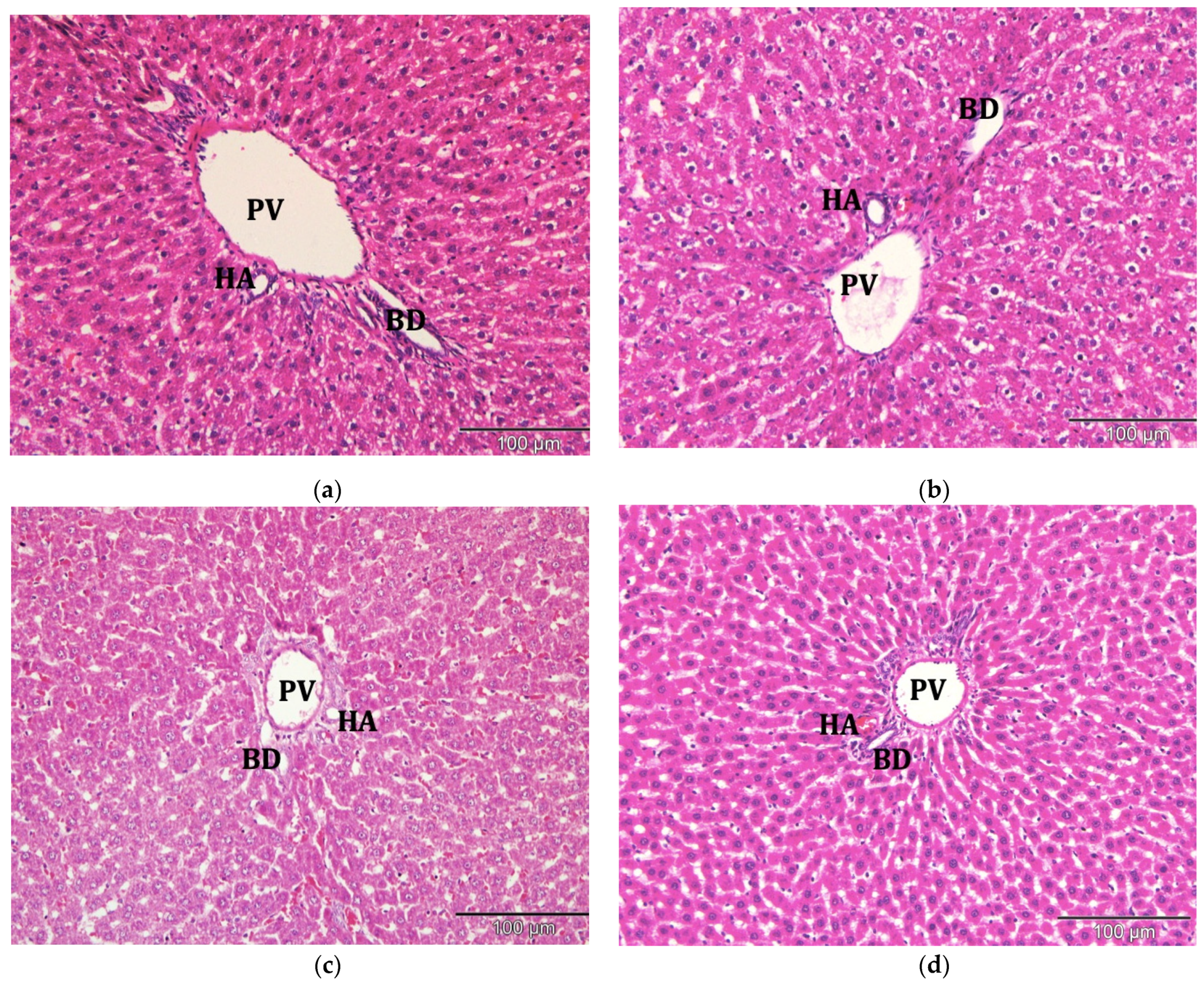
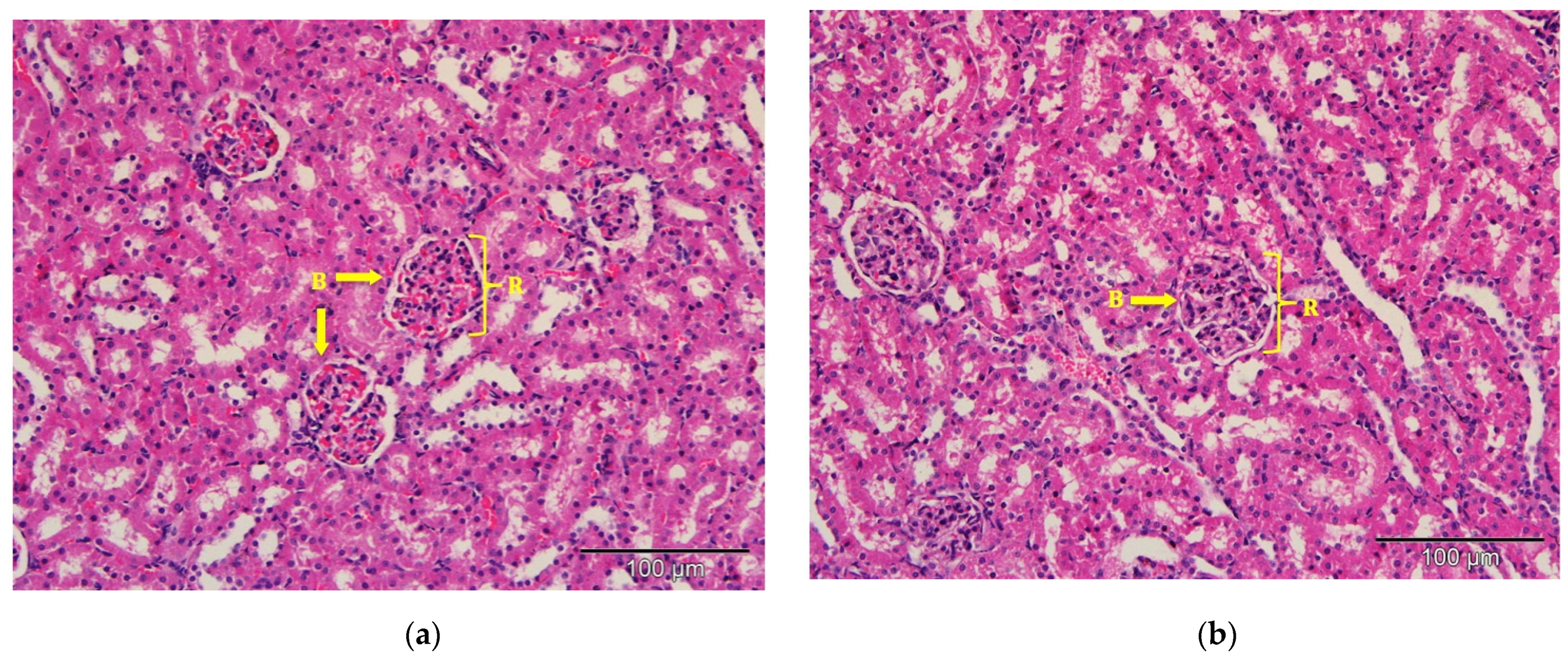
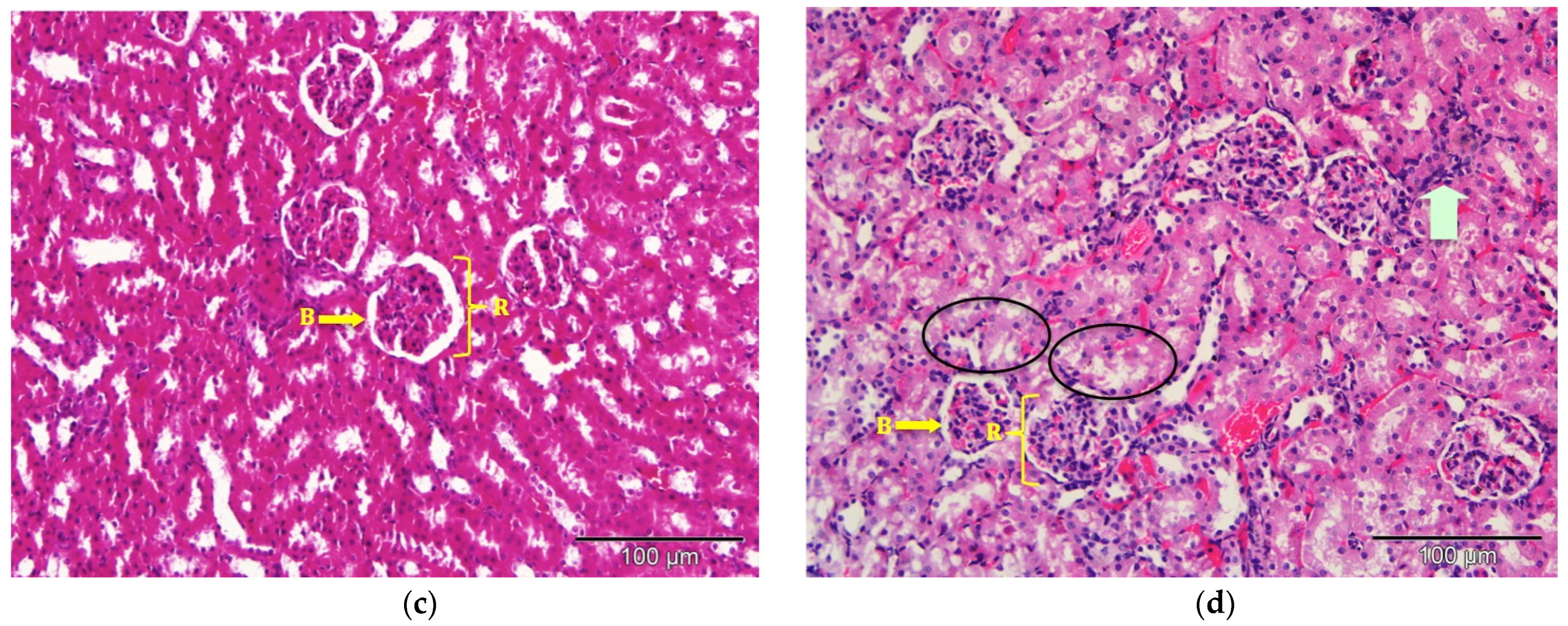
2.9. Histological Observation under the Light Microscope
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Central Composite Design
4.2.1. Statistical Analysis
4.2.2. Verification of the Models
4.3. Preparation of Solid Lipid Nanoparticles
4.4. Size, Zeta Potential and Polydispersity Index Analysis
4.5. Drug Entrapment Efficiency
4.6. Transmission Electron Microscopy
4.7. Differential Scanning Calorimetry
4.8. In Vitro Release Study
4.9. Short-Term Stability Test
4.10. Animal
4.11. Blood Sample Collection and Plasma Preparation
4.12. Ultra-Performance Liquid Chromatography (UPLC)
4.13. Plasma Protein Precipitation for Determination of Acyclovir Concentration
4.14. Pharmacokinetic Parameters
4.15. Collection of Organ Samples
4.16. Tissue Processing
4.17. Light Microscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Elion, G.B. Acyclovir: Discovery, mechanism of action, and selectivity. J. Med. Virol. 1993, 41, 2–6. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Resistance of Herpes Simplex Viruses to Nucleoside Analogues: Mechanisms, Prevalence, and Management. Antimicrob. Agents Chemother. 2010, 55, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Kubbinga, M.; Nguyen, M.A.; Staubach, P.; Teerenstra, S.; Langguth, P. The Influence of Chitosan on the Oral Bioavailability of Acyclovir-a Comparative Bioavailability Study in Humans. Pharm. Res. 2015, 32, 2241–2249. [Google Scholar] [CrossRef] [Green Version]
- Poirier, J.-M.; Radembino, N.; Jaillon, P. Determination of Acyclovir in Plasma by Solid-Phase Extraction and Column Liquid Chromatography. Ther. Drug Monit. 1999, 21, 129–133. [Google Scholar] [CrossRef]
- Wald, A.; Benedetti, J.; Davis, G.; Remington, M.; Winter, C.; Corey, L. A randomized, double-blind, comparative trial comparing high- and standard-dose oral acyclovir for first-episode genital herpes infections. Antimicrob. Agents Chemother. 1994, 38, 174–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, H.; Adam, S.K.; Othman, F.; Shamsuddin, A.F.; Basir, R. Antiviral Nanodelivery Systems: Current Trends in Acyclovir Administration. J. Nanomater. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Rao, L.; Yu, G.; Cook, T.R.; Chen, X.; Huang, F. Supramolecular cancer nanotheranostics. Chem. Soc. Rev. 2021, 50, 2839–2891. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.; Bello, R.O.; Adam, S.K.; Alias, E.; Affandi, M.M.R.; Shamsuddin, A.F.; Basir, R. Acyclovir-Loaded Solid Lipid Nanoparticles: Optimization, Characterization and Evaluation of Its Pharmacokinetic Profile. Nanomaterials 2020, 10, 1785. [Google Scholar] [CrossRef] [PubMed]
- Basit, H.M.; Amin, M.C.I.M.; Ng, S.-F.; Katas, H.; Shah, S.U.; Khan, N.R. Formulation and Evaluation of Microwave-Modified Chitosan-Curcumin Nanoparticles—A Promising Nanomaterials Platform for Skin Tissue Regeneration Applications Following Burn Wounds. Polymers 2020, 12, 2608. [Google Scholar] [CrossRef]
- Akbarzadeh, I.; Yaraki, M.T.; Ahmadi, S.; Chiani, M.; Nourouzian, D. Folic acid-functionalized niosomal nanoparticles for selective dual-drug delivery into breast cancer cells: An in-vitro investigation. Adv. Powder Technol. 2020, 31, 4064–4071. [Google Scholar] [CrossRef]
- Ghafelehbashi, R.; Akbarzadeh, I.; Yaraki, M.T.; Lajevardi, A.; Fatemizadeh, M.; Saremi, L.H. Preparation, physicochemical properties, in vitro evaluation and release behavior of cephalexin-loaded niosomes. Int. J. Pharm. 2019, 569, 118580. [Google Scholar] [CrossRef]
- Patel, D.; Sawant, K.K. Oral Bioavailability Enhancement of Acyclovir by Self-Microemulsifying Drug Delivery Systems (SMEDDS). Drug Dev. Ind. Pharm. 2007, 33, 1318–1326. [Google Scholar] [CrossRef]
- Chetoni, P.; Rossi, S.; Burgalassi, S.; Monti, D.; Mariotti, S.; Saettone, M.F. Comparison of Liposome-Encapsulated Acyclovir with Acyclovir Ointment: Ocular Pharmacokinetics in Rabbits. J. Ocul. Pharmacol. Ther. 2004, 20, 169–177. [Google Scholar] [CrossRef]
- Schwarz, J.C.; Klang, V.; Karall, S.; Mahrhauser, D.; Resch, G.P.; Valenta, C. Optimisation of multiple W/O/W nanoemulsions for dermal delivery of aciclovir. Int. J. Pharm. 2012, 435, 69–75. [Google Scholar] [CrossRef]
- Ghosh, S.; Jhanji, V.; Lamoureux, E.; Taylor, H.; Vajpayee, R.B. Acyclovir Therapy in Prevention of Recurrent Herpetic Keratitis Following Penetrating Keratoplasty. Am. J. Ophthalmol. 2008, 145, 198–202. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Majithiya, R.J.; Umrethia, M.L.; Murthy, R.S.R. Design and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSciTech 2006, 7, E172–E177. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Seyfoddin, A.; Sherwin, T.; Patel, D.; McGhee, C.N.; Rupenthal, I.; Taylor, J.A.; Al-Kassas, R. Ex vivo and In vivo Evaluation of Chitosan Coated Nanostructured Lipid Carriers for Ocular Delivery of Acyclovir. Curr. Drug Deliv. 2016, 13, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.H.; Keck, C. Challenges and solutions for the delivery of biotech drugs—a review of drug nanocrystal technology and lipid nanoparticles. J. Biotechnol. 2004, 113, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Muchow, M.; Maincent, P.; Müller, R.H. Lipid Nanoparticles with a Solid Matrix (SLN®, NLC®, LDC®) for Oral Drug Delivery. Drug Dev. Ind. Pharm. 2008, 34, 1394–1405. [Google Scholar] [CrossRef]
- Müller, R.; Runge, S.; Ravelli, V.; Mehnert, W.; Thünemann, A.; Souto, E. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int. J. Pharm. 2006, 317, 82–89. [Google Scholar] [CrossRef]
- Ugazio, E.; Cavalli, R.; Gasco, M.R. Incorporation of cyclosporin A in solid lipid nanoparticles (SLN). Int. J. Pharm. 2002, 241, 341–344. [Google Scholar] [CrossRef]
- Hu, F.Q.; Wu, M.Z.; Yuan, H.; Zhang, H.H. A novel preparation of solid lipid nanoparticles with cyclosporin A for prolonged drug release. Die Pharm. Int. J. Pharm. Sci. 2004, 59, 683–685. [Google Scholar]
- Peng, X.; Fang, X. Development and optimization of solid lipid nanoparticle formulation for ophthalmic delivery of chloramphenicol using a Box-Behnken design. Int. J. Nanomed. 2011, 6, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Savic Gajic, I.; Savic, I.; Boskov, I.; Žerajić, S.; Markovic, I.; Gajic, D. Optimization of Ultrasound-Assisted Extraction of Phenolic Compounds from Black Locust (Robiniae Pseudoacaciae) Flowers and Comparison with Conventional Methods. Antioxidants 2019, 8, 248. [Google Scholar] [CrossRef] [Green Version]
- Gajic, I.S.; Savic, I.; Gajic, D.; Dosic, A. Ultrasound-Assisted Extraction of Carotenoids from Orange Peel Using Olive Oil and Its Encapsulation in Ca-Alginate Beads. Biomolecules 2021, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Zainol, S.; Basri, M.; Bin Basri, H.; Shamsuddin, A.F.; Gani, S.S.A.; Karjiban, R.A.; Abdul-Malek, E. Formulation Optimization of a Palm-Based Nanoemulsion System Containing Levodopa. Int. J. Mol. Sci. 2012, 13, 13049–13064. [Google Scholar] [CrossRef] [PubMed]
- Padhye, S.G.; Nagarsenker, M.S. Simvastatin Solid Lipid Nanoparticles for Oral Delivery: Formulation Development and In vivo Evaluation. Indian J. Pharm. Sci. 2013, 75, 591–598. [Google Scholar] [PubMed]
- Zirak, M.; Pezeshki, A. Effect of Surfactant Concentration on the Particle Size, Stability and Potential Zeta of Beta carotene Nano Lipid Carrier. Int. J. Curr. Microbiol. App. Sci. 2015, 4, 924–932. [Google Scholar]
- Mohtar, N.; Khan, N.A.K.; Darwis, Y. Solid Lipid Nanoparticles of Atovaquone Based on 24 Full-Factorial Design. Iran. J. Pharm. Res. IJPR 2015, 14, 989–1000. [Google Scholar]
- Asasutjarit, R.; Sorrachaitawatwong, C.; Tipchuwong, N.; Pouthai, S. Effect of formulation compositions on particle size and zeta potential of diclofenac sodium-loaded chitosan nanoparticles. Int. J. Pharmacol. Pharm. Sci. 2013, 7, 568–570. [Google Scholar]
- Asasutjarit, R.; Lorenzen, S.-I.; Sirivichayakul, S.; Ruxrungtham, K.; Ruktanonchai, U.; Ritthidej, G.C. Effect of Solid Lipid Nanoparticles Formulation Compositions on Their Size, Zeta Potential and Potential for In Vitro pHIS-HIV-Hugag Transfection. Pharm. Res. 2007, 24, 1098–1107. [Google Scholar] [CrossRef]
- Kakkar, V.; Singh, S.; Singla, D.; Kaur, I.P. Exploring solid lipid nanoparticles to enhance the oral bioavailability of curcumin. Mol. Nutr. Food Res. 2011, 55, 495–503. [Google Scholar] [CrossRef]
- Seyfoddin, A.; Al-Kassas, R. Development of solid lipid nanoparticles and nanostructured lipid carriers for improving ocular delivery of acyclovir. Drug Dev. Ind. Pharm. 2012, 39, 508–519. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Bunjes, H.; Unruh, T. Characterization of lipid nanoparticles by differential scanning calorimetry, X-ray and neutron scattering. Adv. Drug Deliv. Rev. 2007, 59, 379–402. [Google Scholar] [CrossRef] [PubMed]
- Westesen, K.; Siekmann, B.; Koch, M.H. Investigations on the physical state of lipid nanoparticles by synchrotron radiation X-ray diffraction. Int. J. Pharm. 1993, 93, 189–199. [Google Scholar] [CrossRef]
- Reis, S.; Neves, A.R.; Lúcio, M.; Martins, S.; Lima, J. Novel resveratrol nanodelivery systems based on lipid nanoparticles to enhance its oral bioavailability. Int. J. Nanomed. 2013, 8, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Vivek, K.; Reddy, H.; Murthy, R.S.R. Investigations of the effect of the lipid matrix on drug entrapment, in vitro release, and physical stability of olanzapine-loaded solid lipid nanoparticles. AAPS PharmSciTech 2007, 8, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souto, E.; Anselmi, C.; Centini, M.; Müller, R. Preparation and characterization of n-dodecyl-ferulate-loaded solid lipid nanoparticles (SLN®). Int. J. Pharm. 2005, 295, 261–268. [Google Scholar] [CrossRef]
- Kim, J.T.; Barua, S.; Kim, H.; Hong, S.-C.; Yoo, S.-Y.; Jeon, H.; Cho, Y.; Gil, S.; Oh, K.; Lee, J. Absorption Study of Genistein Using Solid Lipid Microparticles and Nanoparticles: Control of Oral Bioavailability by Particle Sizes. Biomol. Ther. 2017, 25, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.; Kumar, A.; Wild, W.; Ferreira, D.; Santos, D.; Forbes, B. Long-term stability, biocompatibility and oral delivery potential of risperidone-loaded solid lipid nanoparticles. Int. J. Pharm. 2012, 436, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Pizzol, C.D.; Filippin-Monteiro, F.B.; Restrepo, J.A.S.; Pittella, F.; Silva, A.H.; de Souza, P.A.; de Campos, A.M.; Creczynski-Pasa, T.B. Influence of Surfactant and Lipid Type on the Physicochemical Properties and Biocompatibility of Solid Lipid Nanoparticles. Int. J. Environ. Res. Public Health 2014, 11, 8581–8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, C.; Müller, R.H. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN) dispersions. Int. J. Pharm. 1998, 168, 221–229. [Google Scholar] [CrossRef]
- Shah, R.M.; Eldridge, D.S.; Palombo, E.; Harding, I. Optimisation and stability assessment of solid lipid nanoparticles using particle size and zeta potential. J. Phys. Sci. 2014, 25, 59–75. [Google Scholar]
- Harde, H.; Das, M.; Jain, S. Solid lipid nanoparticles: An oral bioavailability enhancer vehicle. Expert Opin. Drug Deliv. 2011, 8, 1407–1424. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.P.; Labhasetwar, V.; Amidon, G.L.; Levy, R.J. Gastrointestinal Uptake of Biodegradable Microparticles: Effect of Particle Size. Pharm. Res. 1996, 13, 1838–1845. [Google Scholar] [CrossRef]
- Mahapatro, A.; Singh, D.K. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J. Nanobiotechnol. 2011, 9, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyhers, H.; Ehlers, S.; Hahn, H.; Souto, E.B.; Müller, R.H. Solid lipid nanoparticles (SLN)-effects of lipid composition on in vitro degradation and in vivo toxicity. Die Pharm. 2006, 61, 539–544. [Google Scholar]
- Lu, H.; Han, Y.-J.; Xu, J.-D.; Xing, W.-M.; Chen, J. Proteomic Characterization of Acyclovir-Induced Nephrotoxicity in a Mouse Model. PLoS ONE 2014, 9, e103185. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, M.H.; Webb, D.E.; Balow, J.E.; Straus, S.E. Acyclovir-induced renal failure: Clinical course and histology. Am. J. Med. 1988, 84, 1067–1071. [Google Scholar] [CrossRef] [Green Version]
- Perazella, M.A. Crystal-induced acute renal failure. Am. J. Med. 1999, 106, 459–465. [Google Scholar] [CrossRef]
- Brigden, D.; Rosling, A.E.; Woods, N.C. Renal function after acyclovir intravenous injection. Am. J. Med. 1982, 73, 182–185. [Google Scholar] [CrossRef]
- Thirupathi, G.; Swetha, E.; Narendar, D. Role of Isradipine Loaded Solid Lipid Nanoparticles on the Pharmacodynamic Effect in Rats. Drug Res. 2016, 67, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Uner, M.; Wissing, S.A.; Yener, G.; Müller, R.H. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for application of ascorbyl palmitate. Die Pharm. 2005, 60, 577–582. [Google Scholar]
- Najafi, R.B.; Mostafavi, A.; Tavakoli, N.; Taymouri, S.; Shahraki, M.-M. Preparation and in vitro-in vivo evaluation of acyclovir floating tablets. Res. Pharm. Sci. 2017, 12, 128–136. [Google Scholar] [CrossRef]
- Jain, S.K.; Jain, R.K.; Chourasia, M.K.; Jain, A.K.; Chalasani, K.B.; Soni, V.; Jain, A. Design and development of multivesicular liposomal depot delivery system for controlled systemic delivery of acyclovir sodium. AAPS PharmSciTech 2005, 6, E35–E41. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and Eosin Staining of Tissue and Cell Sections. Cold Spring Harb. Protoc. 2008, 2008, pdb-prot4989. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size, R1 | Equation: 93.92 + 44.30A − 5.93B + 14.38A2 + 51.75B2 − 36.38AB R2 value: 0.9997 p-value: <0.0001 |
| Zeta Potential, R2 | Equation: −40.20 + 2.24A − 1.25B − 0.36A2 − 1.53B2 − 1.25AB R2 value: 0.9620 p-value: 0.0029 |
| Polydispersity Index, R3 | Equation: 0.20 − 0.04A − 0.07B + 0.06A2 + 0.07B2 − 0.048AB R2 value: 0.9983 p-value: <0.0001 |
| Variables | Size | Zeta Potential | PdI | ||||
|---|---|---|---|---|---|---|---|
| F Value | p-Value | F Value | p-Value | F Value | p-Value | ||
| Main Effects | A | 473.19 | <0.0001 | 6.65 | 0.0495 | 27.73 | 0.0033 |
| B | 8.48 | 0.0333 | 2.08 | 0.2084 | 430.11 | <0.0001 | |
| Quadratic Effects | A2 | 433.32 | <0.0001 | 1.47 | 0.2801 | 540.82 | <0.0001 |
| B2 | 5614.36 | <0.0001 | 27.08 | 0.0035 | 735.67 | <0.0001 | |
| Interaction Effect | AB | 1594.82 | <0.0001 | 10.38 | 0.0234 | 194.09 | <0.0001 |
| Responses | Predicted | Observed |
|---|---|---|
| Particle Size (nm) | 100.00 | 104.89 |
| Polydispersity Index | 0.22 | 0.21 |
| Zeta Potential (mV) | −40.01 | −37.00 |
| Drug-Free SLN | Acyclovir-Loaded SLN | |||||||
|---|---|---|---|---|---|---|---|---|
| Freshly Prepared | 1 Month | 2 Month | 3 Month | Freshly Prepared | 1 Month | 2 Month | 3 Month | |
| Size (nm) | ||||||||
| 4 °C | 104.89 ± 5.53 | 104.11 ± 5.90 | 106.85 ± 3.90 | 106.95 ± 4.42 | 108.68 ± 1.03 | 105.05 ± 0.72 | 106.78 ± 1.79 | 108.33 ± 1.28 |
| 25 °C | 102.28 ± 3.42 | 103.33 ± 2.63 | 102.68 ± 1.04 | 106.85 ± 1.53 | 111.38 ± 3.76 | 113.05 ± 1.79 | ||
| 40 °C | 255.70 ± 8.73 | 292.62 ±14.68 | 330.55 ± 9.73 | 128.43 ± 5.19 | 141.43 ±10.53 | 622.98 ±17.17 | ||
| PdI | ||||||||
| 4 °C | 0.21 ± 0.01 | 0.22 ± 0.03 | 0.22 ± 0.01 | 0.21 ± 0.01 | 0.22 ± 0.03 | 0.20 ± 0.02 | 0.21 ± 0.02 | 0.21 ± 0.01 |
| 25 °C | 0.20 ± 0.01 | 0.20 ± 0.01 | 0.20 ± 0.02 | 0.20 ± 0.01 | 0.20 ± 0.02 | 0.21 ± 0.01 | ||
| 40 °C | 0.33 ± 0.02 | 0.43 ± 0.02 | 0.46 ± 0.01 | 0.29 ± 0.02 | 0.37 ± 0.04 | 0.35 ± 0.02 | ||
| Zeta Potential (mV) | ||||||||
| 4 °C | −37.00 ± 0.89 | −38.13 ± 0.85 | −37.88 ± 1.36 | −35.13 ± 2.21 | −33.45 ± 0.78 | −32.88 ± 1.01 | −34.60 ± 1.28 | −33.98 ± 0.87 |
| 25 °C | −35.28 ± 0.94 | −35.23 ± 1.07 | −36.05 ± 1.54 | −33.45 ± 0.93 | −33.50 ± 1.41 | −34.93 ± 1.31 | ||
| 40 °C | −25.50 ± 0.81 | −27.08 ± 1.19 | −25.23 ± 1.04 | −37.00 ± 1.01 | −25.48 ± 1.56 | −26.38 ± 0.76 | ||
| Parameters | Commercial Acyclovir Suspension | Acyclovir-Loaded SLN |
|---|---|---|
| Cmax (ng/mL) | 303.50 ± 26.70 | 818.67 ± 66.02 |
| Tmax (h) | 1.00 ± 0.00 | 1.00 ± 0.00 |
| AUC0–24 (h.ng.mL−1) | 1243.75 ± 125.90 | 5759.00 ± 346.40 |
| AUC0–∞ (h.ng.mL−1) | 1341.67 ± 133.40 | 6783.14 ± 313.80 |
| Ke (h−1) | 0.37 ± 0.05 | 0.15 ± 0.02 |
| t1/2 (h) | 2.06 ± 0.29 | 5.53 ± 0.99 |
| Independent Variables | Coded Levels | ||||
|---|---|---|---|---|---|
| Axial (−α) | Low | Centre | High | Axial (+α) | |
| Compritol 888 ATO (mg) | 117.16 | 200.00 | 400.00 | 600.00 | 682.84 |
| Tween 80 (% w/w) | 0.17 | 1.00 | 3.00 | 5.00 | 5.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, H.; Adam, S.K.; Alias, E.; Meor Mohd Affandi, M.M.R.; Shamsuddin, A.F.; Basir, R. Central Composite Design for Formulation and Optimization of Solid Lipid Nanoparticles to Enhance Oral Bioavailability of Acyclovir. Molecules 2021, 26, 5432. https://doi.org/10.3390/molecules26185432
Hassan H, Adam SK, Alias E, Meor Mohd Affandi MMR, Shamsuddin AF, Basir R. Central Composite Design for Formulation and Optimization of Solid Lipid Nanoparticles to Enhance Oral Bioavailability of Acyclovir. Molecules. 2021; 26(18):5432. https://doi.org/10.3390/molecules26185432
Chicago/Turabian StyleHassan, Haniza, Siti Khadijah Adam, Ekram Alias, Meor Mohd Redzuan Meor Mohd Affandi, Ahmad Fuad Shamsuddin, and Rusliza Basir. 2021. "Central Composite Design for Formulation and Optimization of Solid Lipid Nanoparticles to Enhance Oral Bioavailability of Acyclovir" Molecules 26, no. 18: 5432. https://doi.org/10.3390/molecules26185432
APA StyleHassan, H., Adam, S. K., Alias, E., Meor Mohd Affandi, M. M. R., Shamsuddin, A. F., & Basir, R. (2021). Central Composite Design for Formulation and Optimization of Solid Lipid Nanoparticles to Enhance Oral Bioavailability of Acyclovir. Molecules, 26(18), 5432. https://doi.org/10.3390/molecules26185432