
Exploring EZH2-Proteasome Dual-Targeting Drug Discovery through a Computational Strategy to Fight Multiple Myeloma


 , ,
, ,  and
and 
Abstract
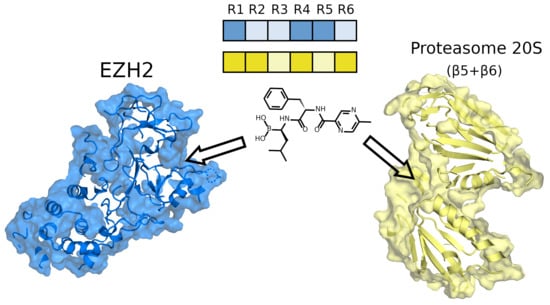
:
1. Introduction
2. Results
2.1. An Overview of the Chemical Space of Known EZH2 and P20S Inhibitors
2.2. Comparison of Binding Pockets in EZH2 and P20S
2.3. Molecular Docking of EZH2 and P20S Inhibitor Datasets to Select Dual-Binding Compounds
2.3.1. Validation of the Molecular Docking Calculations
2.3.2. Construction of a Predictive Model for EZH2 and P20S Inhibitors
2.3.3. Interactions of the Selected Docking Hits
2.4. Construction of QSAR Models Using Machine Learning to Predict Dual-Targeting Inhibitors against P20S and EZH2
2.5. Molecular Dynamics Studies to Investigate Dual-Inhibitor Hits Produced from Molecular Docking
3. Conclusions
4. Methods
4.1. P20S and EZH2 Ligands Dataset Compilation and Preparation
4.2. Preparation of P20S and EZH2 Protein Structures
4.3. Structural Analysis of Inhibitors and Protein Pockets
4.4. Molecular Docking (Validation, Parameters, and Simulations)
4.5. Prediction Performance Metrics for Molecular Docking
4.6. Building of the Machine Learning (Decision Tree) Classification Model
4.7. Topology, Systems Setup, and Molecular Dynamics Simulation Protocols
4.8. Analysis of the Molecular Dynamics Simulations
4.9. Data Management, Data Analysis Plots and Figures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1106. [Google Scholar] [CrossRef] [Green Version]
- Branagan, A.; Lei, M.; Lou, U.; Raje, N. Current treatment strategies for multiple myeloma. J. Oncol. Pract. 2020, 16, 5–14. [Google Scholar] [CrossRef]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S.V.U.M. Dual or multi-targeting inhibitors: The next generation anticancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- East, S.P.; Silver, L.L. Multitarget ligands in antibacterial research: Progress and opportunities. Expert Opin. Drug Discov. 2012, 8, 143–156. [Google Scholar] [CrossRef]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef]
- Alcaro, S.; Bolognesi, M.L.; García-Sosa, A.T.; Rapposelli, S. Editorial: Multi-target-directed ligands (MTDL) as challenging research tools in drug discovery: From design to pharmacological evaluation. Front. Chem. 2019, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Guedes, R.A.; Aniceto, N.; Andrade, M.A.P.; Salvador, J.A.R.; Guedes, R.C. Chemical patterns of proteasome inhibitors: Lessons learned from two decades of drug design. Int. J. Mol. Sci. 2019, 20, 5326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, L.J.; Irvine, A.E. Targeting the ubiquitin proteasome system in haematological malignancies. Blood Rev. 2013, 27, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Cai, C.-Y.; Assaraf, Y.G.; Guo, H.-Q.; Cui, Q.; Wei, L.; Huang, J.-J.; Ashby, C.R.; Chen, Z.-S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updat. 2019, 48, 100663. [Google Scholar] [CrossRef] [PubMed]
- Fricker, L.D. Proteasome inhibitor drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.; Chiecchio, L.; Smith, E.M.; Boyd, K.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef]
- Pawlyn, C.; Bright, M.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Rothe, N.; Curry, E.; Zeller, C.; Liber, D.; Stronach, E.; Gabra, H.; Ghaem-Maghami, S.; Brown, R. Chromatin H3K27me3/H3K4me3 histone marks define gene sets in high-grade serous ovarian cancer that distinguish malignant, tumour-sustaining and chemo-resistant ovarian tumour cells. Oncogene 2013, 32, 4586–4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Yu, J.; Rhodes, D.R.; Tomlins, S.A.; Cao, X.; Chen, G.; Mehra, R.; Wang, X.; Ghosh, D.; Shah, R.B. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007, 67, 10657–10663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvà, M.-L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.-C.; Baumer, K.; Le Bitoux, M.-A.; Marino, D.; Cironi, L.; et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.W.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013, 125, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef] [Green Version]
- Rizq, O.; Mimura, N.; Oshima, M.; Saraya, A.; Koide, S.; Kato, Y.; Aoyama, K.; Nakajima-Takagi, Y.; Wang, C.; Chiba, T.; et al. Dual inhibition of EZH2 and EZH1 sensitizes PRC2-dependent tumors to proteasome inhibition. Clin. Cancer Res. 2017, 23, 4817–4830. [Google Scholar] [CrossRef] [Green Version]
- Ashtiani, M.; Salehzadeh-Yazdi, A.; Razaghi-Moghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A systematic survey of centrality measures for protein-protein interaction networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.U. Polypharmacology—Foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Albertini, C.; Naldi, M.; Petralla, S.; Strocchi, S.; Grifoni, D.; Monti, B.; Bartolini, M.; Bolognesi, M. From combinations to single-molecule polypharmacology—cromolyn-ibuprofen conjugates for alzheimer’s disease. Molecules 2021, 26, 1112. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Aittokallio, T. Improving the efficacy-safety balance of polypharmacology in multi-target drug discovery. Expert Opin. Drug Discov. 2018, 13, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Krieger, V.; Groll, M.; Osko, J.; Reßing, N.; Ahlert, H.; Borkhardt, A.; Kurz, T.; Christianson, D.W.; Hauer, J.; et al. Discovery of the first-in-class dual histone deacetylase-proteasome inhibitor. J. Med. Chem. 2018, 61, 10299–10309. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X.; Xue, J.; Liu, L.; Liang, T.; Li, W.; Yang, X.; Hou, X.; Fang, H. Discovery of peptide boronate derivatives as histone deacetylase and proteasome dual inhibitors for overcoming bortezomib resistance of multiple myeloma. J. Med. Chem. 2020, 63, 4701–4715. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Harshbarger, W.D.; Robles, O.; Krysiak, J.; Hull, K.G.; Cho, S.W.; Richardson, R.D.; Yang, Y.; Garcia, A.; Spiegelman, L.; et al. A strategy for dual inhibition of the proteasome and fatty acid synthase with belactosin C-orlistat hybrids. Bioorg. Med. Chem. 2017, 25, 2901–2916. [Google Scholar] [CrossRef] [Green Version]
- Bratkowski, M.; Yang, X.; Liu, X. An evolutionarily conserved structural platform for PRC2 inhibition by a class of Ezh2 inhibitors. Sci. Rep. 2018, 8, 9092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, J.; Heinneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkow, G.; Chari, A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Wang, S.; Balius, T.; Singh, I.; Levit, A.; Moroz, Y.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Jia, L.; Du, F.; Dong, X.; Sun, W.; Wang, L.; Chen, G. Design, synthesis and biological activities of pyrrole-3-carboxamide derivatives as EZH2 (enhancer of zeste homologue 2) inhibitors and anticancer agents. N. J. Chem. 2020, 44, 2247–2255. [Google Scholar] [CrossRef]
- Huang, K.; Sun, R.; Chen, J.; Yang, Q.; Wang, Y.; Zhang, Y.; Xie, K.; Zhang, T.; Li, R.; Zhao, Q.; et al. A novel EZH2 inhibitor induces synthetic lethality and apoptosis in PBRM1-deficient cancer cells. Cell Cycle 2020, 19, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, F.; Konze, K.D.; Meslamani, J.; Ma, A.; Brown, P.J.; Zhou, M.-M.; Arrowsmith, C.H.; Kaniskan, H.; Vedadi, M.; et al. Structure-activity relationship studies for enhancer of zeste homologue 2 (EZH2) and enhancer of zeste homologue 1 (EZH1) inhibitors. J. Med. Chem. 2016, 59, 7617–7633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aier, I.; Varadwaj, P.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Du, D.; Yang, R.; Tao, H.; Zhang, H. Identification and assessments of novel and potent small-molecule inhibitors of EED–EZH2 interaction of polycomb repressive complex 2 by computational methods and biological evaluations. Chem. Pharm. Bull. 2020, 68, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aier, I.; Raj, U. Exploring the role of EZH2 (PRC2) as epigenetic target. In Proceedings of the 2016 International Conference on Bioinformatics and Systems Biology (BSB), Allahabad, India, 4–6 March 2016. [Google Scholar] [CrossRef]
- Raj, U.; Kumar, H.; Gupta, S.; Varadwaj, P. Identification of Novel Inhibitors for Disrupting EZH2-EED Interactions Involved in Cancer Epigenetics: An In-Silico Approach. Curr. Proteom. 2016, 13, 313–321. [Google Scholar] [CrossRef]
- Li, A.; Sun, H.; Du, L.; Wu, X.; Cao, J.; You, Q.; Li, Y. Discovery of novel covalent proteasome inhibitors through a combination of pharmacophore screening, covalent docking, and molecular dynamics simulations. J. Mol. Model. 2014, 20, 2515. [Google Scholar] [CrossRef]
- Di Giovanni, C.; Ettari, R.; Sarno, S.; Rotondo, A.; Bitto, A.; Squadrito, F.; Altavilla, D.; Schirmeister, T.; Novellino, E.; Grasso, S.; et al. Identification of noncovalent proteasome inhibitors with high selectivity for chymotrypsin-like activity by a multistep structure-based virtual screening. Eur. J. Med. Chem. 2016, 121, 578–591. [Google Scholar] [CrossRef]
- Yadav, D.; Mishra, B.N.; Khan, F. Quantitative structure-activity relationship and molecular docking studies on human proteasome inhibitors for anticancer activity targeting NF-κB signaling pathway. J. Biomol. Struct. Dyn. 2020, 38, 3621–3632. [Google Scholar] [CrossRef]
- Kazi, A.; Lawrence, H.; Guida, W.C.; McLaughlin, M.L.; Springett, G.M.; Berndt, N.; Yip, R.M.L.; Sebti, S.M. Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle 2009, 8, 1940–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arba, M.; Nur-Hidayat, A.; Surantaadmaja, S.I.; Tjahjono, D.H. Pharmacophore-based virtual screening for identifying β5 subunit inhibitor of 20S proteasome. Comput. Biol. Chem. 2018, 77, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Uysal, S.; Soyer, Z.; Saylam, M.; Tarikogullari, A.H.; Yilmaz, S.; Kirmizibayrak, P.B. Design, synthesis and biological evaluation of novel naphthoquinone-4-aminobenzensulfonamide/carboxamide derivatives as proteasome inhibitors. Eur. J. Med. Chem. 2021, 209, 112890. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Mehrotra, M.; Gupta, D. Support vector machine based prediction of P. falciparum proteasome inhibitors and development of focused library by molecular docking. Comb. Chem. High Throughput Screen. 2011, 14, 898–907. [Google Scholar] [CrossRef]
- Sánchez-Cruz, N.; Medina-Franco, J.L. Epigenetic target profiler: A web server to predict epigenetic targets of small molecules. J. Chem. Inf. Model. 2021, 61, 1550–1554. [Google Scholar] [CrossRef]
- Cruciani, G.; Crivori, P.; Carrupt, P.A.; Testa, B. Molecular fields in quantitative structure-permeation relationships: The VolSurf approach. J. Mol. Struct. THEOCHEM 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Moorthy, N.S.H.N.; Ramos, M.J.; Fernandes, P.A. Structural analysis of structurally diverse α-glucosidase inhibitors for active site feature analysis. J. Enzym. Inhib. Med. Chem. 2012, 27, 649–657. [Google Scholar] [CrossRef]
- Arba, M.; Nur-Hidayat, A.; Ruslin; Yusuf, M.; Sumarlin; Hertadi, R.; Wahyudi, S.T.; Surantaadmaja, S.I.; Tjahjono, D.H. Molecular modeling on porphyrin derivatives as β5 subunit inhibitor of 20S proteasome. Comput. Biol. Chem. 2018, 74, 230–238. [Google Scholar] [CrossRef]
- Zhang, S.; Shi, Y.; Jin, H.; Liu, Z.; Zhang, L.; Zhang, L. Covalent complexes of proteasome model with peptide aldehyde inhibitors MG132 and MG101: Docking and molecular dynamics study. J. Mol. Model. 2009, 15, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.S.F.F.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.; Uhalte, E.C.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; A James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 23 July 2021).
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Neudert, G.; Klebe, G. fconv: Format conversion, manipulation and feature computation of molecular data. Bioinformatics 2011, 27, 1021–1022. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Lin, Z.; Van Gunsteren, W.F. Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J. Chem. Theory Comput. 2011, 7, 1237–1243. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLIFs Similarity | IC50 (nM) | |||
|---|---|---|---|---|
| EZH2 | P20S | EZH2 | P20S | |
| CHEMBL3794075 | 0.900 | 0.917 | unknown | 26.24 |
| CHEMBL3771372 | 0.900 | 0.917 | 10.00 | unknown |
| Class | N (Test) | Precision | Recall | F1 | Overall Accuracy | |
|---|---|---|---|---|---|---|
| EZH2 | actives | 29 | 0.84 | 0.90 | 0.87 | 0.76 |
| moderate actives | 6 | 0.60 | 0.43 | 0.50 | ||
| inactives | 7 | 0.50 | 0.50 | 0.50 | ||
| P20S | actives | 74 | 0.84 | 0.93 | 0.88 | 0.77 |
| moderate actives | 14 | 0.44 | 0.44 | 0.44 | ||
| inactives | 18 | 0.83 | 0.36 | 0.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrada, F.G.A.; Miccoli, S.; Aniceto, N.; García-Sosa, A.T.; Guedes, R.C. Exploring EZH2-Proteasome Dual-Targeting Drug Discovery through a Computational Strategy to Fight Multiple Myeloma. Molecules 2021, 26, 5574. https://doi.org/10.3390/molecules26185574
Estrada FGA, Miccoli S, Aniceto N, García-Sosa AT, Guedes RC. Exploring EZH2-Proteasome Dual-Targeting Drug Discovery through a Computational Strategy to Fight Multiple Myeloma. Molecules. 2021; 26(18):5574. https://doi.org/10.3390/molecules26185574
Chicago/Turabian StyleEstrada, Filipe G. A., Silvia Miccoli, Natália Aniceto, Alfonso T. García-Sosa, and Rita C. Guedes. 2021. "Exploring EZH2-Proteasome Dual-Targeting Drug Discovery through a Computational Strategy to Fight Multiple Myeloma" Molecules 26, no. 18: 5574. https://doi.org/10.3390/molecules26185574
APA StyleEstrada, F. G. A., Miccoli, S., Aniceto, N., García-Sosa, A. T., & Guedes, R. C. (2021). Exploring EZH2-Proteasome Dual-Targeting Drug Discovery through a Computational Strategy to Fight Multiple Myeloma. Molecules, 26(18), 5574. https://doi.org/10.3390/molecules26185574