
GPCR Pharmacological Profiling of Aaptamine from the Philippine Sponge Stylissa sp. Extends Its Therapeutic Potential for Noncommunicable Diseases



Abstract
:1. Introduction
2. Results and Discussion
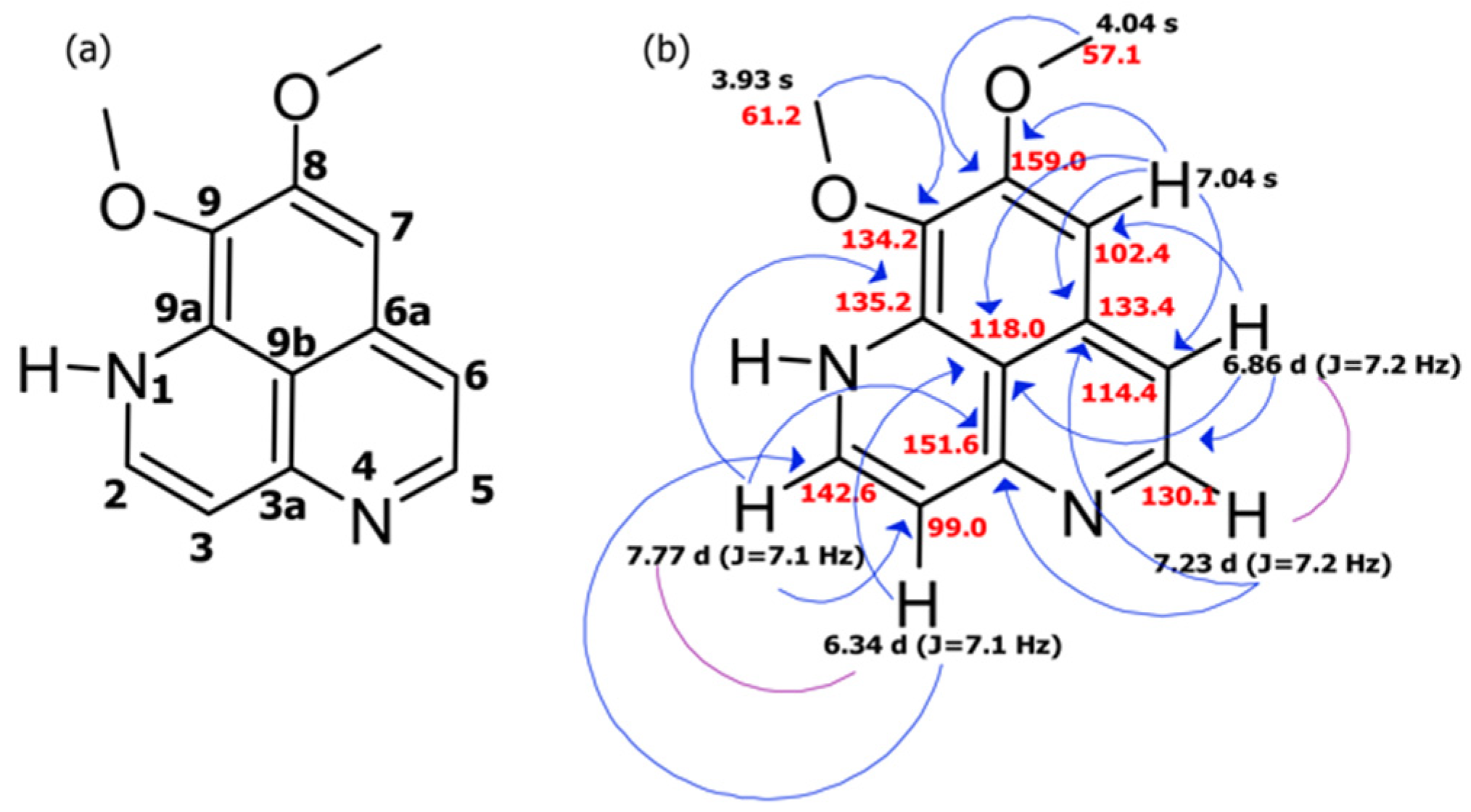
2.1. Collection, Isolation, and Structure Elucidation
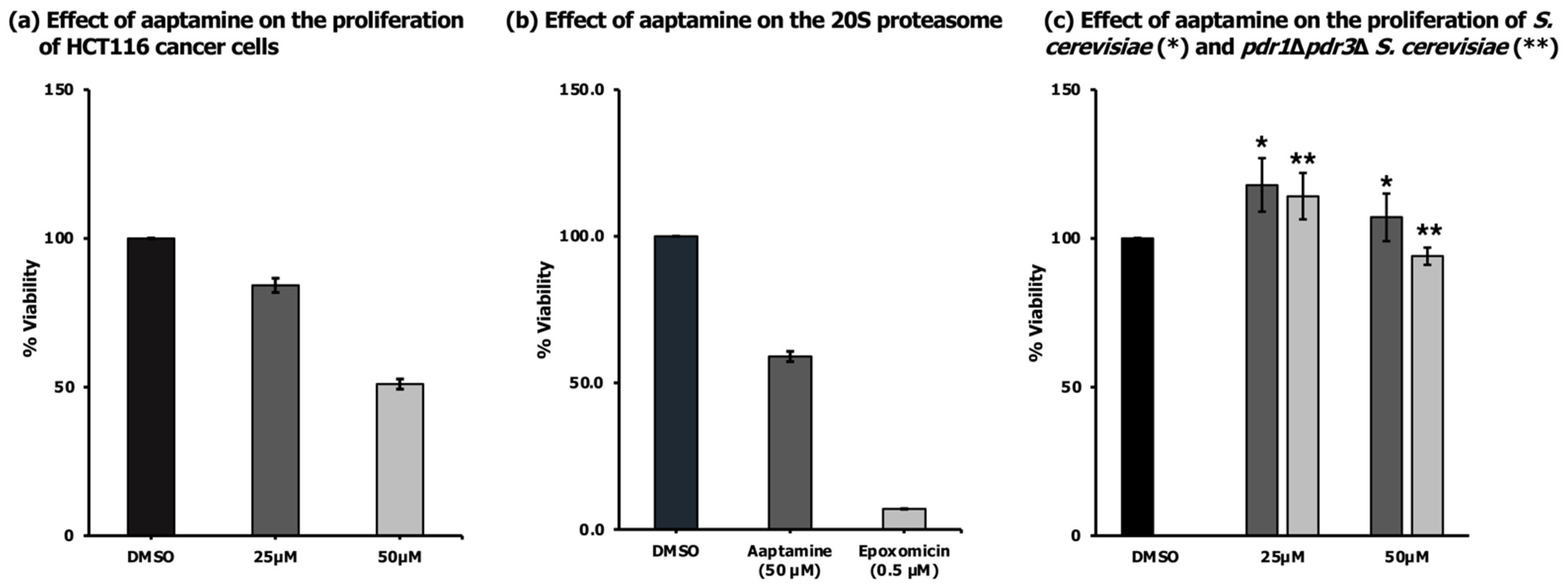
2.2. Antiproliferative Activity of Aaptamine
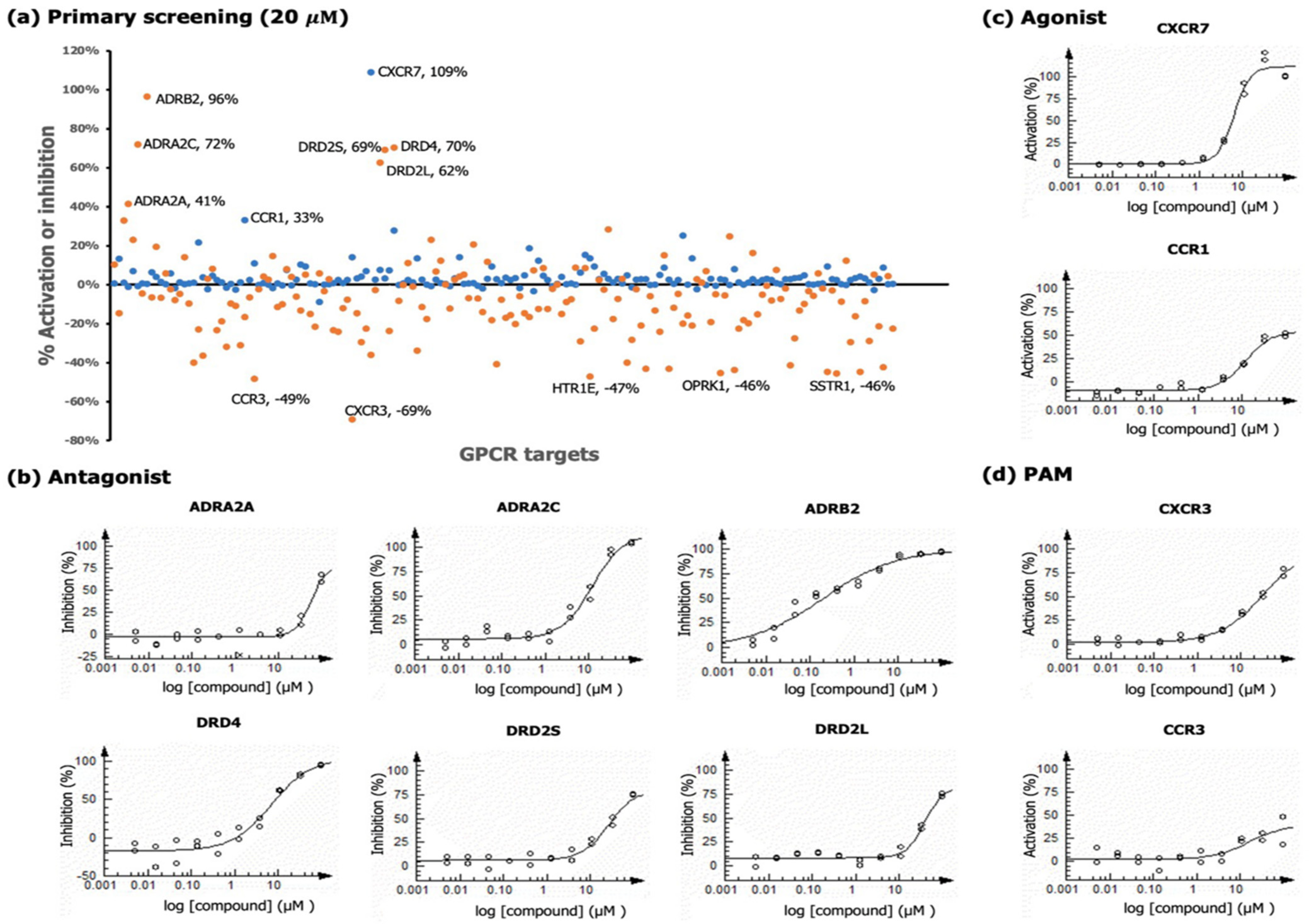
2.3. GPCR Analysis
2.3.1. Chemokine Receptors
2.3.2. Adrenergic Receptors
2.3.3. Dopamine Receptors
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Collection and Sample Preparation
3.3. Extraction and Isolation
3.4. Bioassays
3.4.1. Antiproliferative Activity against Cancer Cells
3.4.2. Proteasome Assay
3.4.3. Test for Activity against Saccharomyces cerevisiae
3.4.4. S. Cerevisiae pdr1pdr3 Double Deletion Strain
3.4.5. GPCR Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nakamura, H.; Kobayashi, J.; Ohizumi, Y.; Hirata, Y. Isolation and structure of aaptamine a novel heteroaromatic substance possessing α-blocking activity from the sea sponge Aaptos aaptos. Tetrahedron Lett. 1982, 23, 5555–5558. [Google Scholar] [CrossRef]
- Bergquist, P.R.; Cambie, R.C.; Kernan, M.R. Aaptamine, a taxonomic marker for sponges of the order hadromerida. Biochem. Syst. Ecol. 1991, 19, 289–290. [Google Scholar] [CrossRef]
- Calcul, L.; Longeon, A.; Al Mourabit, A.; Guyot, M.; Bourguet-Kondracki, M.L. Novel alkaloids of the aaptamine class from an indonesian marine sponge of the genus Xestospongia. Tetrahedron 2003, 59, 6539–6544. [Google Scholar] [CrossRef]
- Larghi, E.L.; Bohn, M.L.; Kaufman, T.S. Aaptamine and related products. Their isolation, chemical syntheses, and biological activity. Tetrahedron 2009, 65, 4257–4282. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Yamanokuchi, R.; Yoshitomi, M.; Sato, K.; Ikeda, T.; Rotinsulu, H.; Mangindaan, R.E.P.; de Voogd, N.J.; van Soest, R.W.M.; Yokosawa, H. Aaptamine, an alkaloid from the sponge Aaptos suberitoides, functions as a proteasome inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 3341–3343. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Milan-Lobo, L.; Che, T.; Ferwerda, M.; Lambu, E.; McIntosh, N.L.; Whistler, J.L. Identification of the first marine-derived opioid receptor “Balanced” agonist with a signaling profile that resembles the endorphins. ACS Chem. Neurosci. 2017, 8, 473–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Zhou, Q.; Labroska, V. G protein-coupled receptors: Structure-and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Sriram, K.; Gorr, M.W.; Wiley, S.Z.; Michkov, A.; Salmer, N.C.; Chinn, A.M. GPCRomics: An approach to discover GPCR drug targets. Trends Pharmacol. Sci. 2019, 40, 378–387. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schith, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets, and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Budreviciute, A.; Damiati, S.; Sabir, D.; Onder, K.; Schuller-Goetzberg, P.; Plakys, G.; Katileviciute, A.; Khoja, S.; Kodzius, R. Management and prevention strategies for noncommunicable diseases (NCDs) and their risk factors. Front. Public Health 2020. [Google Scholar] [CrossRef] [PubMed]
- Dalesio, N.M.; Barreto-Ortiz, S.F.; Pluznick, J.L.; Berkowitz, D.E. Olfactory, taste, and photo sensory receptors in non-sensory organs: It just makes sense. Front. Physiol. 2018, 9, 1673. [Google Scholar] [CrossRef] [Green Version]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Ohizumi, Y.; Kajiwara, A.; Nakamura, H.; Kobayashi, J. α-Adrenoceptor blocking action of aaptamine, a novel marine natural product, in vascular smooth muscle. J. Pharm. Pharmacol. 1984, 36, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Kitamura, K.; Nagai, K.; Nakao, Y.; Fusetani, N.; Van Soest, R.W.M.; Matsunaga, S. Carteramine A, an inhibitor of neutrophil chemotaxis, from the marine sponge Stylissa carteri. Tetrahedron Lett. 2007, 48, 2127–2129. [Google Scholar] [CrossRef]
- Diers, J.A.; Bowling, J.J.; Duke, S.O.; Wahyuono, S.; Kelly, M.; Hamann, M.T. Zebra Mussel antifouling activity of the marine natural product Aaptamine and Analogs. Mar. Biotechnol. 2006, 8, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Bertics, P.J.; Kozio-White, C.; Gavala, M.L.; Wiepz, G.J. 11-Signal Transduction. Middleton’s Allergy 2014, 8, 184–202. [Google Scholar] [CrossRef]
- Romagnani, P.; Lasagni, L.; Annunziato, F.; Serio, M.; Romagnani, S. CXC chemokines: The regulatory link between inflammation and angiogenesis. Trends Immunol. 2004, 25, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Arimont, M.; Hoffmann, C.; de Graaf, C.; Leurs, R. Chemokine receptor crystal structures: What can be learnt from them? Mol. Pharmacol. 2019, 96, 765–777. [Google Scholar] [CrossRef]
- Wells, T.N.; Power, C.A.; Shaw, J.P.; Proudfoot, A.E.I. Chemokine blockers. Therapeutics in the making? Trends Pharmacol. Sci. 2006, 27, 41–47. [Google Scholar] [CrossRef]
- De Menthon, M.; Lavalley, M.P.; Maldini, C.; Guillevin, L.; Mahr, A. HLA-B51/B5 and the risk of Behcet’s disease: A systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheumatol. 2009, 61, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Revankar, S.G.; Sobel, J.D. Chromoblastomycosis. In The Merck Manual; Professional Edition; Merck & Co.: Kenilworth, NJ, USA, 2021; Available online: http://www.merckmanuals.com/professional/infectious_diseases/fungi/chromoblastomycosis.html (accessed on 16 January 2021).
- Laurent, V.; Gurard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef]
- Coperchini, F.; Croce, L.; Marin, M.; Chiovato, L.; Rotondi, M. Role of chemokine receptors in thyroid cancer and immunotherapy. Endocr.-Relat. Cancer 2019, 26, R465–R478. [Google Scholar] [CrossRef] [Green Version]
- Gudowska-Sawczuk, M.; Kudelski, J.; Mroczko, B. The role of chemokine receptor CXCR3 and its ligands in renal cell carcinoma. Int. J. Mol. Sci. 2020, 21, 8582. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, W.; Shen, J. CXCR7 targeting and its major disease relevance. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, N. The multiple faces of CXCL12 (SDF-1α) in the regulation of immunity during health and disease. J. Leukoc. Biol. 2010, 88, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Crawford, D.; Tsuchihashi, T.; Behrens, T.W.; Srivastava, D. The chemokine receptor CXCR7 functions to regulate cardiac valve remodeling. Dev. Dyn. 2011, 240, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. Chemokines as mediators of neovascularization. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1928–1936. [Google Scholar] [CrossRef] [Green Version]
- Sekiguchi, H.; Kuroyanagi, T.; Rhainds, D.; Kobayashi, K.; Kobayashi, Y.; Ohno, H.; Heveker, N.; Akaji, K.; Fujii, N.; Oishi, S. Structure-activity relationship study of cyclic pentapeptide ligands for atypical chemokine receptor 3 (ACKR3). J. Med. Chem. 2018, 61, 3745–3751. [Google Scholar] [CrossRef]
- Gravel, S.; Malouf, C.; Boulais, P.E.; Berchiche, Y.A.; Oishi, S.; Fujii, N.; Leduc, R.; Sinnett, D.; Heveker, N. The peptidomimetic CXCR4 antagonist TC14012 recruits β-arrestin to CXCR7: Roles of receptor domains. J. Biol. Chem. 2010, 285, 37939–37943. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.; Sakmar, T.P.; Rafii, S.; Ding, B. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Al-Awadhi, F.H.; Gao, B.; Rezaei, M.A.; Kwan, J.C.; Li, C.; Ye, T.; Paul, V.J.; Luesch, H. Discovery, synthesis, pharmacological profiling, and biological characterization of brintonamides A–E, novel dual protease and GPCR modulators from a marine cyanobacterium. J. Med. Chem. 2018, 61, 6364–6378. [Google Scholar] [CrossRef]
- Liang, X.; Lou, D.; Yan, J.L.; Rezaei, M.; Salvador-Reyes, L.; Gunasekera, S.; Li, C.; Ye, T.; Paul, V.; Luesch, H. Discovery of amantamide, a selective CXCR7 agonist from marine cyanobacteria. Org. Lett. 2019, 21, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Uto-Konomi, A.; McKibben, B.; Wirtz, J.; Sato, Y.; Takano, A.; Nanki, T.; Suzuki, S. CXCR7 agonists inhibit the function of CXCL12 by down-regulation of CXCR4. Biochem. Biophys. Res. Commun. 2013, 431, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ye, Y.; Long, P.; Zhao, S.; Zhang, L.; Yanni, A. Silencing of CXCR4 and CXCR7 expression by RNA interference suppresses human endometrial carcinoma growth in vivo. Am. J. Transl. Res. 2017, 9, 1896–1904. [Google Scholar] [PubMed]
- Ciccarelli, M.; Sorriento, D.; Coscioni, E.; Iaccarino, G.; Santulli, G. Adrenergic receptors. In Endocrinology of the Heart in Health and Disease; Schisler, J.C., Lang, C.H., Willis, M.S., Eds.; Academic Press: Durham, NC, USA, 2017; pp. 285–315. [Google Scholar] [CrossRef]
- Simões, R.; Ferreira, J.; Barbosa, A.P.; Mascarenhas, M.R.; Bicho, M. P02. Beta-2 adrenergic receptor (ADRB2) gene polymorphisms as risk factors for reduced bone mineral density. Rev. Port. Endocrinol. Diabetes Metab. 2016, 11, 12–13. [Google Scholar] [CrossRef] [Green Version]
- Giovannitti, J.A.; Thoms, S.M.; Crawford, J.J. Alpha-2 Adrenergic receptor agonists: A review of current clinical applications. Anesth. Prog. 2015, 62, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Hieble, J.P. Adrenergic receptors. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Durham, NC, USA, 2009; Volume 10, pp. 135–139. [Google Scholar] [CrossRef]
- Gribble, F.M. α2A-adrenergic receptors and type 2 diabetes. N. Engl. J. Med. 2010, 362, 361–362. [Google Scholar] [CrossRef]
- Martel, J.C.; McArthur, S.G. Dopamine receptor subtypes, physiology and pharmacology: New ligands and concepts in schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, W.; Chen, K.; Zhao, Y.; Ye, F.; Tang, X. Serum epidermal growth factor is low in schizophrenia and not affected by antipsychotics alone or combined with electroconvulsive therapy. Front. Psychiatry 2020, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Zuk, J.; Bartuzi, D.; Matosiuk, D.; Kaczor, A.A. Preferential coupling of dopamine D2S and D2L receptor isoforms with Gi1 and Gi2 proteins-in silico study. Int. J. Mol. Sci. 2020, 21, 436. [Google Scholar] [CrossRef] [Green Version]
- Amato, D.; Kruyer, A.; Samaha, A.N.; Heinz, A. Hypofunctional dopamine uptake and antipsychotic treatment-resistant schizophrenia. Front. Psychiatry 2019, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, S.; Grizenko, N.; Joober, R. Dopamine genes and attention-deficit hyperactivity disorder: A review. J. Psychiatry Neurosci. 2003, 28, 27–38. [Google Scholar]
- Mayseless, N.; Uzefovsky, F.; Shalev, I.; Ebstein, R.P.; Shamay-Tsoory, S.G. The association between creativity and 7R polymorphism in the dopamine receptor D4 gene (DRD4). Front. Hum. Neurosci. 2013, 7, 502. [Google Scholar] [CrossRef] [Green Version]
- Di Ciano, P.; Grandy, D.K.; Le Foll, B. Dopamine D4 receptors in psychostimulant addiction. Adv. Pharmacol. 2014, 301–321. [Google Scholar] [CrossRef] [Green Version]
- Yet, L. Pyrazoles. Compr. Heterocycl. Chem. III 2008, 1–141. [Google Scholar] [CrossRef]
- Elsadek, L.; Matthews, J.; Nishimura, S.; Nakatani, T.; Ito, A.; Gu, T.; Lou, D.; Salvador-Reyes, L.; Paul, V.; Kakeya, H.; et al. Genomic and targeted approaches unveil the cell membrane as a major target of the antifungal cytotoxin amantelide A. ChemBioChem 2021, 22, 1790–1799. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| C/H No. | δc, Type | δH (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 2 | 142.6 CH | 7.77 d (J = 7.1) | H: 3 | C: 3, 3a, 9a |
| 3 | 99.0 CH | 6.34 d (J = 7.1) | H: 2 | C: 2, 9b |
| 3a | 151.6 C | |||
| 5 | 130.1 CH | 7.23 d (J = 7.2) | H: 6 | C: 3a, 6, 6a |
| 6 | 114.4 CH | 6.86 d (J = 7.2) | H: 5 | C: 5, 7, 9b |
| 6a | 133.4 C | |||
| 7 | 102.4 CH | 7.04 s | C: 6, 6a, 8, 9b | |
| 8 | 159.0 C | |||
| 9 | 134.2 C | |||
| 9a | 135.2 C | |||
| 9b | 118.0 C | |||
| 8-OMe | 57.1 CH3 | 4.04 s | C: 8 | |
| 9-OMe | 61.2 CH3 | 3.93 s | C: 9 |
| Assay Format | GPCR | Primary Screen | Dose-Response Analysis | ||
|---|---|---|---|---|---|
| Maximal Response, % Inhibition or Activation | Result Type | IC50 or EC50 Value, µM | Maximal Response, % Inhibition or Activation | ||
| Antagonist a | ADRB2 | 96 | IC50 | 0.20 | 96.5 |
| DRD4 | 70 | IC50 | 6.9 | 94.5 | |
| ADRA2C | 72 | IC50 | 11.9 | 103.9 | |
| DRD2S | 69 | IC50 | 25.7 | 74.4 | |
| DRD2L | 62 | IC50 | 38.7 | 73.7 | |
| ADRA2A | 41 | IC50 | 56.1 | 63.4 | |
| Agonist | CXCR7 | 109 | EC50 | 6.2 | 122.57 |
| CCR1 | 33 | EC50 | 11.8 | 50.2 | |
| PAM a | CCR3 | 49 | EC50 | 16.2 | 33.0 |
| CXCR3 | 69 | EC50 | 31.8 | 74.6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luyao, H.; Luesch, H.; Uy, M. GPCR Pharmacological Profiling of Aaptamine from the Philippine Sponge Stylissa sp. Extends Its Therapeutic Potential for Noncommunicable Diseases. Molecules 2021, 26, 5618. https://doi.org/10.3390/molecules26185618
Luyao H, Luesch H, Uy M. GPCR Pharmacological Profiling of Aaptamine from the Philippine Sponge Stylissa sp. Extends Its Therapeutic Potential for Noncommunicable Diseases. Molecules. 2021; 26(18):5618. https://doi.org/10.3390/molecules26185618
Chicago/Turabian StyleLuyao, Harmie, Hendrik Luesch, and Mylene Uy. 2021. "GPCR Pharmacological Profiling of Aaptamine from the Philippine Sponge Stylissa sp. Extends Its Therapeutic Potential for Noncommunicable Diseases" Molecules 26, no. 18: 5618. https://doi.org/10.3390/molecules26185618
APA StyleLuyao, H., Luesch, H., & Uy, M. (2021). GPCR Pharmacological Profiling of Aaptamine from the Philippine Sponge Stylissa sp. Extends Its Therapeutic Potential for Noncommunicable Diseases. Molecules, 26(18), 5618. https://doi.org/10.3390/molecules26185618