
New Flavone C-Glycosides from Scleranthus perennis and Their Anti-Collagenase Activity





Abstract
:1. Introduction
2. Results and Discussion
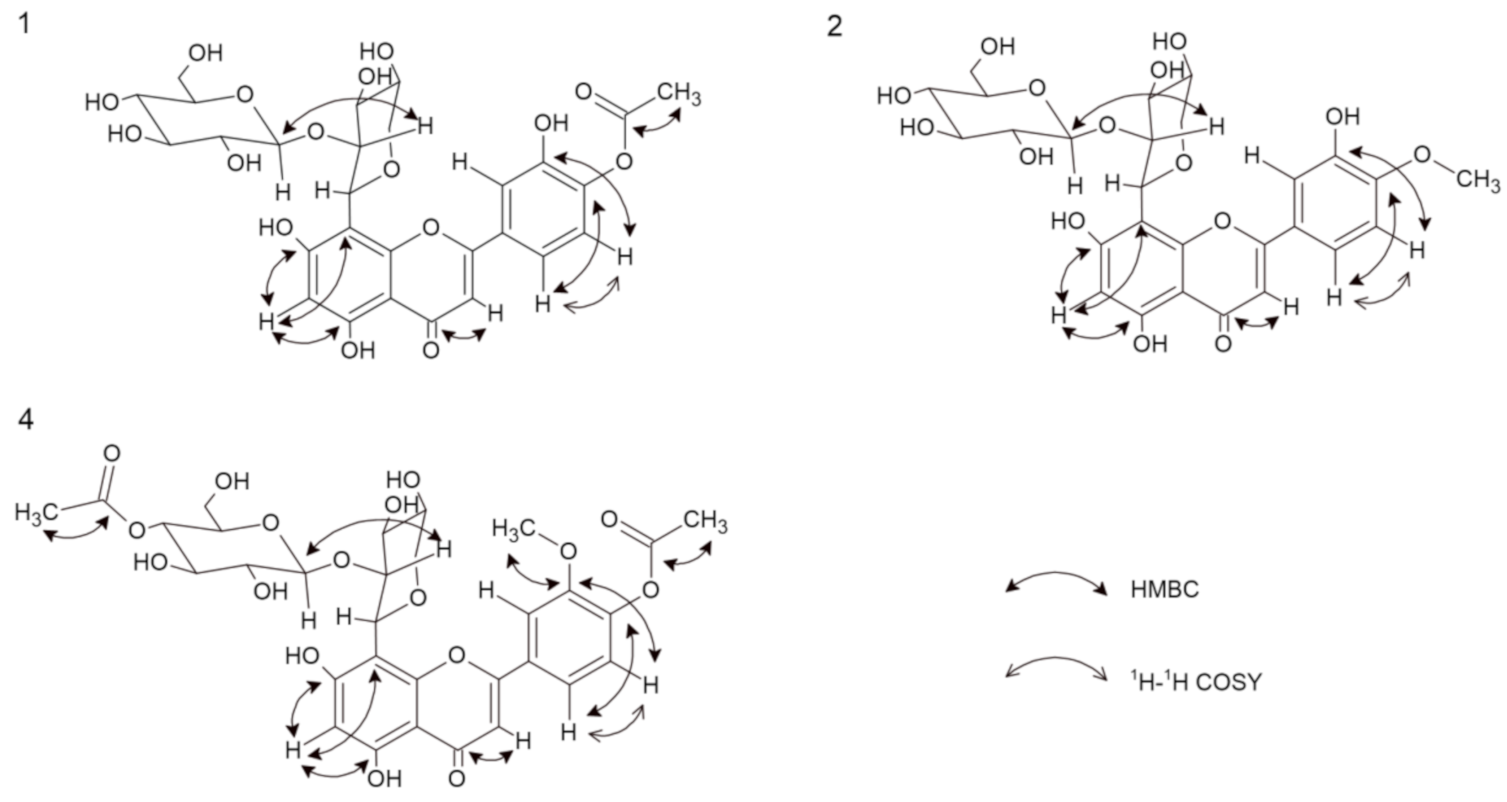
2.1. 5,7,3′-Trihydroxy-4′-acetoxyflavone-8-C-β-d-xylopyranoside-2″-O-glucoside (1)
2.2. 5,7,3′-Trihydroxy-4′-methoxyflavone-8-C-β-d-xylopyranoside-2″-O-glucoside (2)
2.3. 5,7-Dihydroxy-3′-methoxy-4′-acetoxyflavone-8-C-β-d-xylopyranoside-2″-O-glucoside (3)
2.4. 5,7-Dihydroxy-3′-methoxy-4′-acetoxyflavone-8-C-β-d-xylopyranoside-2″-O-(4″′-acetoxy)-glucoside (4)
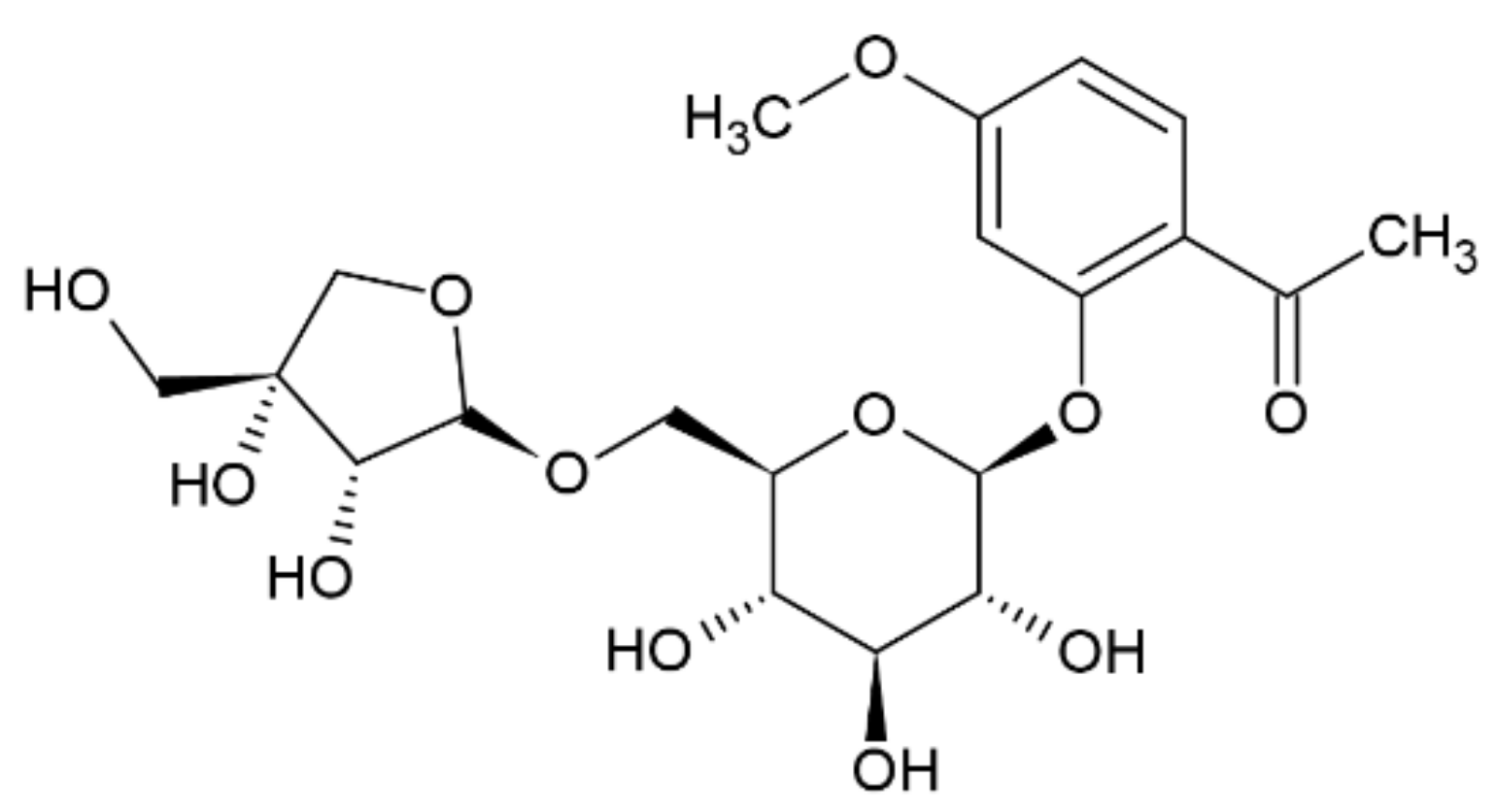
2.5. Apiopaenonside (5)
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis
3.5. Isolates
3.6. In Vitro Collagenase Inhibition Assay
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| NMR | nuclear magnetic resonance |
| COSY | correlation spectroscopy |
| HSQC | heteronuclear single quantum coherence |
| HMBC | heteronuclear multiple bond correlation |
| DEPT | distortionless enhancement by polarization transfer |
| IR | infrared spectroscopy |
| UV | ultraviolet radiation |
| UV-Vis | ultraviolet-visible spectroscopy |
| ESI | electrospray ionization |
| IC50 | median inhibitory concentration |
| MS | mass spectrometer |
| LC-MS | liquid chromatography–mass spectrometry |
| HPLC | high-performance liquid chromatography |
| HRESIMS | high-resolution electrospray ionization mass spectrometry |
| TLC | thin-layer chromatography |
| Glc | glucose |
| Xyl | xylose |
| NaOAc | sodium acetate |
| NaOMe | sodium methoxide |
| AlCl3 | aluminum chloride |
| NaCl | sodium chloride |
| CaCl2 | calcium chloride |
| FALGPA | N-[3-(2-Furyl)acryloyl]-leu-gly-Pro-Ala |
| EGCG | epigallocatechin gallate |
| TOF | time-of-flight |
| CD3OD | deuterated methanol |
| DMSO | dimethyl sulfoxide |
| CC | column chromatography |
| MeOH | methanol |
| Et2O | diethyl ether |
| EtOAc | ethyl acetate |
| n-BuOH | n-butanol |
| UPW | ultra-pure water |
| ACN | acetonitrile |
| mp | melting point |
| SD | standard deviation |
| ANOVA | analysis of variance |
| PHS | post-hydrolyzed solution |
References
- Smissen, R.D.; Garnock-Jones, P.J. Relationships, classification and evolution of Scleranthus (Caryophyllaceae) as inferred from analysis of morphological characters. Bot. J. Linn. Soc. 2002, 140, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Sell, P.D. Scleranthus (Caryophyllaceae). In Flora Europaea, 1st ed.; Tutin, T.G., Heywood, V.H., Burges, N.A., Valentine, D.H., Walters, S.M., Webb, D.A., Eds.; Cambridge University Press: London, UK, 1964; pp. 148–149. [Google Scholar]
- Yayli, N.; Seymen, H.; Baltaci, C. Flavone C-glycosides from Scleranthus uncinatus. Phytochemistry 2001, 58, 607–610. [Google Scholar] [CrossRef]
- Yayli, N.; Baltaci, C.; Genç, H.; Terzioǧlu, S. Phenolic and flavone C-glycosides from Scleranthus uncinatus. Pharm. Biol. 2002, 40, 369–373. [Google Scholar] [CrossRef]
- Zdraveva, P. Chemical compounds in butanol extracts of Scleranthus perennis L., Caryophyllaceae. In Proceedings of the 4th Conference on Medicinal and Aromatic Plants of South-East European Countries, Iasi, Romania, 28–31 May 2006. [Google Scholar]
- Zdraveva, P.; Assenov, I. Phytochemical study of Scleranthus perennis L. (Caryophyllaceae). Pharmacia 1997, 44, 7–10. [Google Scholar]
- Zdraveva, P.; Gevrenova, R.; Dimitrova, B. Phenolic compounds of Scleranthus annuus L. (Caryophyllaceae). In Proceedings of the 3rd Conference on Medicinal and Aromatic Plants of Southeast European Countries, Nitra, Slovak Republic, 5–8 September 2004. [Google Scholar]
- Svensson, L. An estimate of pollen carryover by ants in a natural population of Scleranthus perennis L. (Caryophyllaceae). Oecologia 1985, 66, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Jakimiuk, K.; Strawa, J.W.; Granica, S.; Tomczyk, M. Flavonoids from the aerial parts of Scleranthus perennis. In Proceedings of the T20 PSE Conference Liverpool 2020 on Contemporary Natural Products Discovery Research, Liverpool, UK, 6 March 2020. [Google Scholar]
- Jakimiuk, K.; Wink, M.; Tomczyk, M. Flavonoids of the Caryophyllaceae. Phytochem. Rev. 2021, 20, 1–40. [Google Scholar] [CrossRef]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Markham, K.R.; Chari, V.M. Carbon-13 NMR spectroscopy of flavonoids. In The Flavonoids; Harborne, J.B., Mabry, T.J., Eds.; Spriger: Boston, MA, USA, 1970. [Google Scholar]
- Agrawal, P.K. Carbon-13 NMR of Flavonoids; Elsevier Science: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Chopin, J.; Dellamonica, G. The Flavonoids; Chapman and Hall: London, UK, 1988. [Google Scholar]
- Wiliams, C.A.; Harborne, J.B. Flavone and flavanol glycosides. In The Flavonoids. Advances in Research Since 1986; Harborne, J.B., Ed.; Chapman and Hall: London, UK, 1994. [Google Scholar]
- Pauli, G.F.; Junior, P. Phenolic glycosides from Adonis aleppica. Phytochemistry 1995, 38, 1245–1250. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, X.; Liu, J.; Kang, L.; Chen, S.; Ma, B.; Guo, B. Quantitative and qualitative analysis of flavonoids and phenolic acids in snow chrysanthemum (Coreopsis tinctoria Nutt.) by HPLC-DAD and UPLC-ESI-QTOF-MS. Molecules 2016, 21, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. Phys. Chem. Chem. Phys. 2014, 16, 1759–1787. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-S.; Wu, Q.; Yin, D.-D.; Feng, C.-Y.; Liu, Z.-A.; Wang, L.-S. Phytochemical variation among the traditional Chinese medicine Mu Dan Pi from Paeonia suffruticosa (tree peony). Phytochemistry 2018, 146, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Ohta, T.; Kawaguchi, A.; Yoshikawa, M. Bioactive constituents of Chinese natural medicines. VI.1) Moutan cortex. (2): Structures and radical scavenging effects of suffruticosides A, B, C, D, and E and galloyl-oxypaeoniflorin. Chem. Pharm. Bull. 2001, 49, 69–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strawa, J.; Wajs-Bonikowska, A.; Jakimiuk, K.; Waluk, M.; Poslednik, M.; Nazaruk, J.; Tomczyk, M. Phytochemical examination of woolly burdock Arctium tomentosum leaves and flower heads. Chem. Nat. Compd. 2020, 56, 345–347. [Google Scholar] [CrossRef]
- Rutkowski, L. Klucz do Oznaczania Roślin Naczyniowych Polski Niżowej; Wydawnictwo Naukowe PWN: Warsaw, Poland, 2006. [Google Scholar]
- Thring, T.S.A.; Hili, P.; Naughton, D.P. Anti-collagenase, anti-elastase and anti-oxidant activities of extracts from 21 plants. BMC Complement. Altern. Med. 2009, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| C No. | 1 | 2 | 4 | |||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |
| 2 | 166.36 | - | 166.21 | - | 164.80 | - |
| 3 | 104.46 | 6.51, s | 104.24 | 6.61, s | 104.01 | 6.60, s |
| 4 | 184.17 | - | 184.25 | - | 182.79 | - |
| 5 | 162.62 | - | 162.82 | - | 161.27 | - |
| 6 | 100.86 | 6.19, s | 100.78 | 6.27, s | 99.47 | 6.21, s |
| 7 | 164.55 | - | 164.56 | - | 163.27 | - |
| 8 | 103.92 | - | 104.38 | - | 102.09 | - |
| 9 | 156.89 | - | 156.95 | - | 155.51 | - |
| 10 | 105.36 | - | 105.48 | - | 106.32 | - |
| -OMe | - | - | 56.69 | 4.01 (s, 3H) | 55.31 | 4.03 (s, 3H) |
| -OAc | 172.98 | - | - | - | 170.29 | - |
| 20.79 | 1.98, s | - | - | 19.08 | 1.93, s | |
| 1′ | 124.16 | - | 124.13 | - | 122.81 | - |
| 2′ | 114.50 | 7.68, s | 111.03 | 7.61, s | 109.71 | 7.66, s |
| 3′ | 146.92 | - | 149.45 | - | 148.09 | - |
| 4′ | 150.90 | - | 151.88 | - | 150.52 | - |
| 5′ | 116.85 | 6.92 (d, J = 8.28) | 117.07 | 6.96 (d, J = 8.28) | 115.41 | 6.96 (d, J = 8.28) |
| 6′ | 120.78 | 7.42 (d, J = 8.28) | 121.84 | 7.50 (d, J = 8.28) | 120.41 | 7.51 (d, J = 8.28) |
| 1′′ | 74.90 | 5.08 (d, 1H, J = 9.54) | 76.02 | 5.10 (d, 1H, J = 9.54) | 71.40 | 5.14 (d, 1H, J = 9.54) |
| 2′′ | 81.66 | 3.99 | 81.36 | 3.85 | 81.52 | 3.87 |
| 3′′ | 75.95 | 3.76 | 77.11 | 3.76 | 73.34 | 3.85 |
| 4′′ | 70.17 | 4.20 | 70.22 | 3.89 | 68.60 | 3.91 |
| 5′′ | 71.92 | 4.06 | 72.09 | 3.87 | 70.58 | 3.90 |
| 1′′′ | 105.89 | 4.29 (d, 1H, J = 7.78) | 105.98 | 4.31 (d, 1H, J = 7.78) | 104.88 | 4.34 (d, 1H, J = 7.78) |
| 2′′′ | 74.90 | 2.91 | 76.02 | 3.15 | 71.40 | 3.33 |
| 3′′′ | 77.76 | 3.01 | 77.98 | 3.19 | 74.64 | 3.37 |
| 4′′′ | 70.67 | 2.93 | 71.82 | 3.17 | 69.68 | 3.23 |
| 5′′′ | 75.57 | 3.15 | 75.88 | 3.27 | 71.40 | 3.38 |
| 6′′ | 64.58 | 3.17 | 63.12 | 3.37 | 61.60 | 3.41 |
| -OAc | - | - | - | - | 171.26 | - |
| - | - | - | - | 19.58 | 1.96, s | |
| Compounds | IC50 a (µM) |
|---|---|
| 1 | 70.24 ± 1.37 |
| 2 | 64.86 ± 1.08 |
| 3 | 48.28 ± 1.05 |
| 4 | 36.06 ± 0.78 |
| 5 | >125 |
| EGCG b | 34.32 ± 0.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakimiuk, K.; Strawa, J.W.; Granica, S.; Tomczyk, M. New Flavone C-Glycosides from Scleranthus perennis and Their Anti-Collagenase Activity. Molecules 2021, 26, 5631. https://doi.org/10.3390/molecules26185631
Jakimiuk K, Strawa JW, Granica S, Tomczyk M. New Flavone C-Glycosides from Scleranthus perennis and Their Anti-Collagenase Activity. Molecules. 2021; 26(18):5631. https://doi.org/10.3390/molecules26185631
Chicago/Turabian StyleJakimiuk, Katarzyna, Jakub W. Strawa, Sebastian Granica, and Michał Tomczyk. 2021. "New Flavone C-Glycosides from Scleranthus perennis and Their Anti-Collagenase Activity" Molecules 26, no. 18: 5631. https://doi.org/10.3390/molecules26185631
APA StyleJakimiuk, K., Strawa, J. W., Granica, S., & Tomczyk, M. (2021). New Flavone C-Glycosides from Scleranthus perennis and Their Anti-Collagenase Activity. Molecules, 26(18), 5631. https://doi.org/10.3390/molecules26185631