
Neurosteroids: Structure-Uptake Relationships and Computational Modeling of Organic Anion Transporting Polypeptides (OATP)1A2

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results
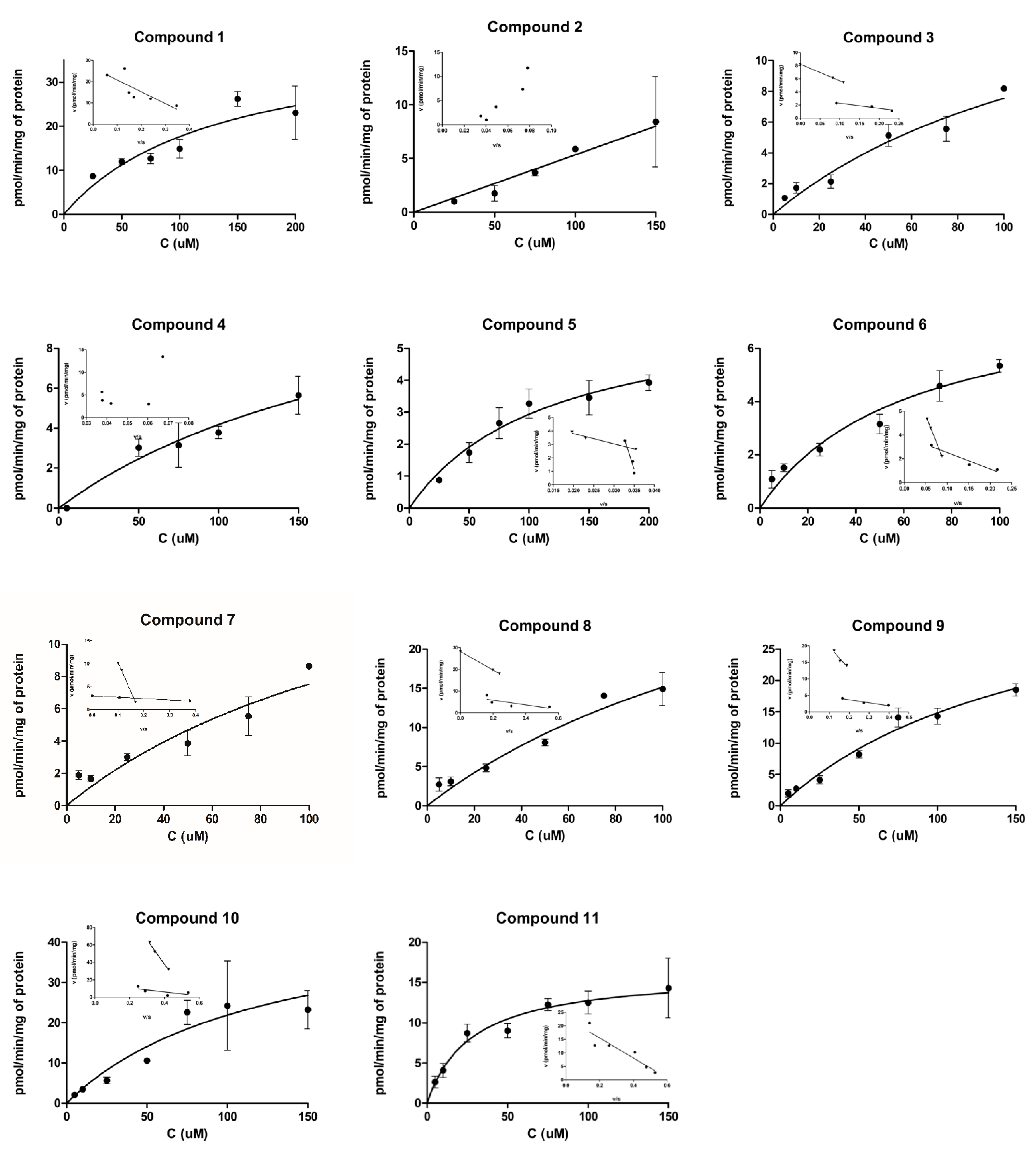
2.1. Cellular Uptake Studies
2.1.1. Cellular Uptake of 3α5β-Pregnanolone (1, CAS Number 128-20-1)
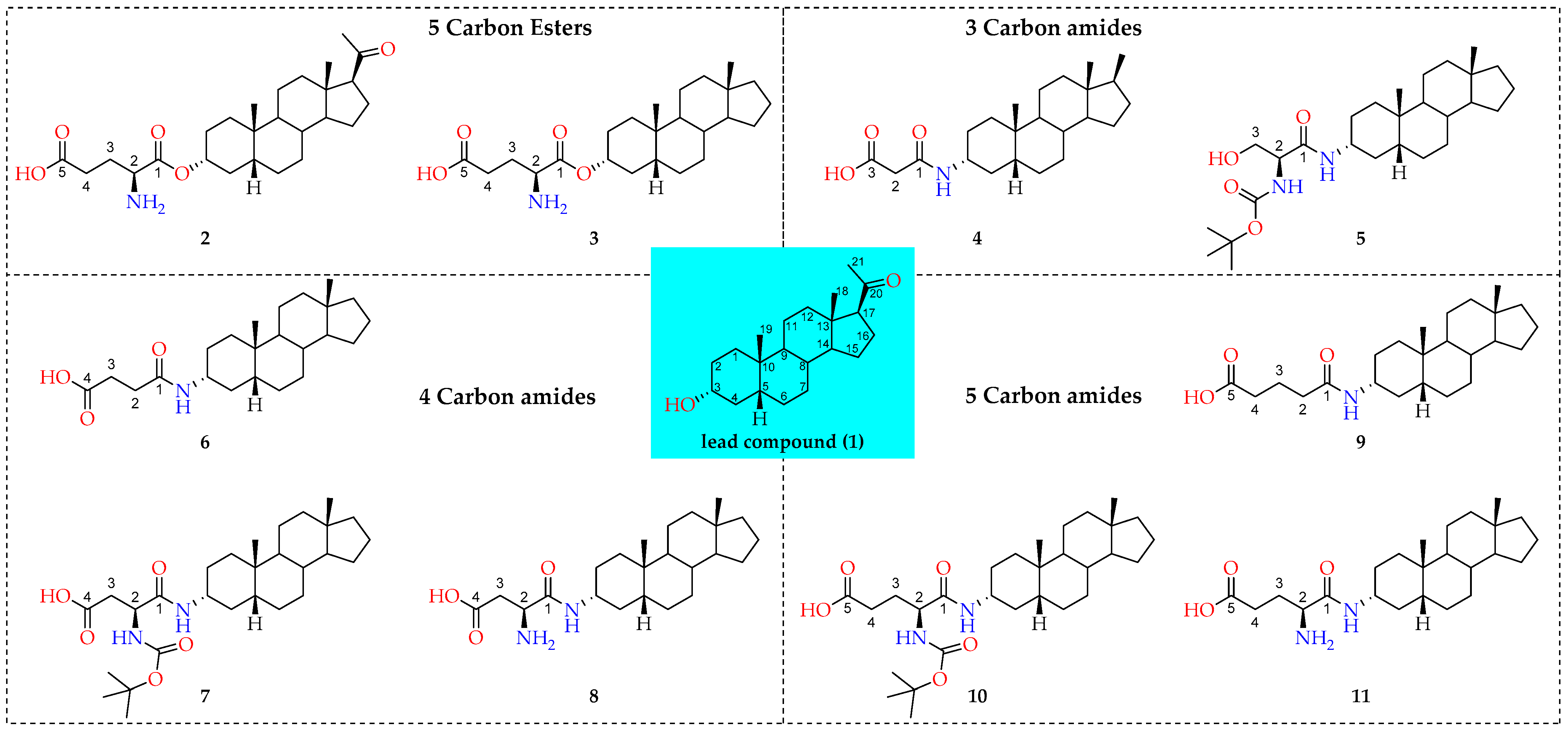
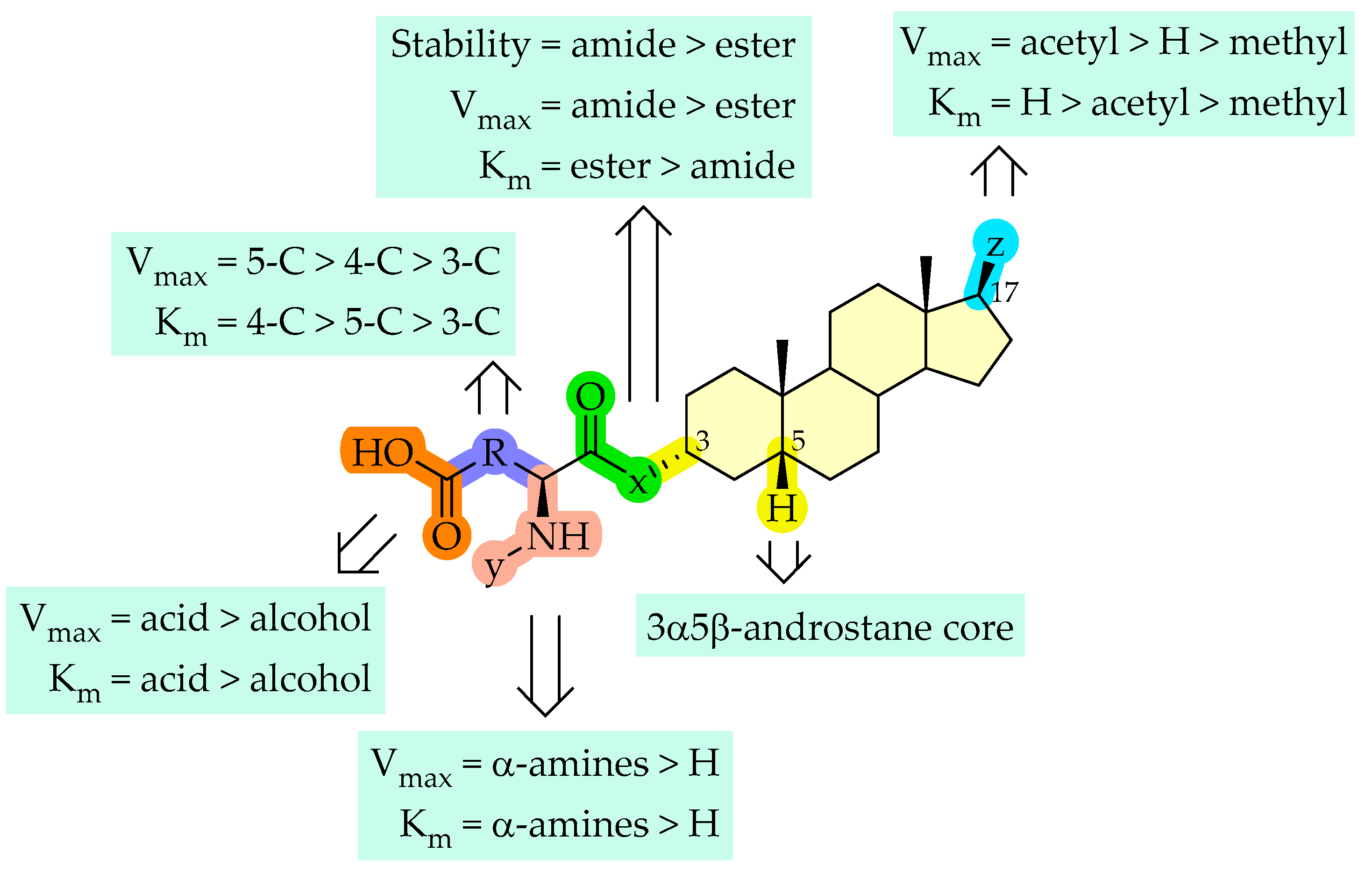
2.1.2. Cellular Uptake of Neurosteroids with Ester Bond (2 and 3)
2.1.3. Cellular Uptake of Neurosteroids Having 3-Carbon Amide Residues (4 and 5)
2.1.4. Cellular Uptake of Neurosteroids Having Medium Length 4-Carbon Amide Residues (6, 7, and 8)
2.1.5. Cellular Uptake of Neurosteroids with Longer Length 5-Carbon Amide Residues (9, 10, and 11)
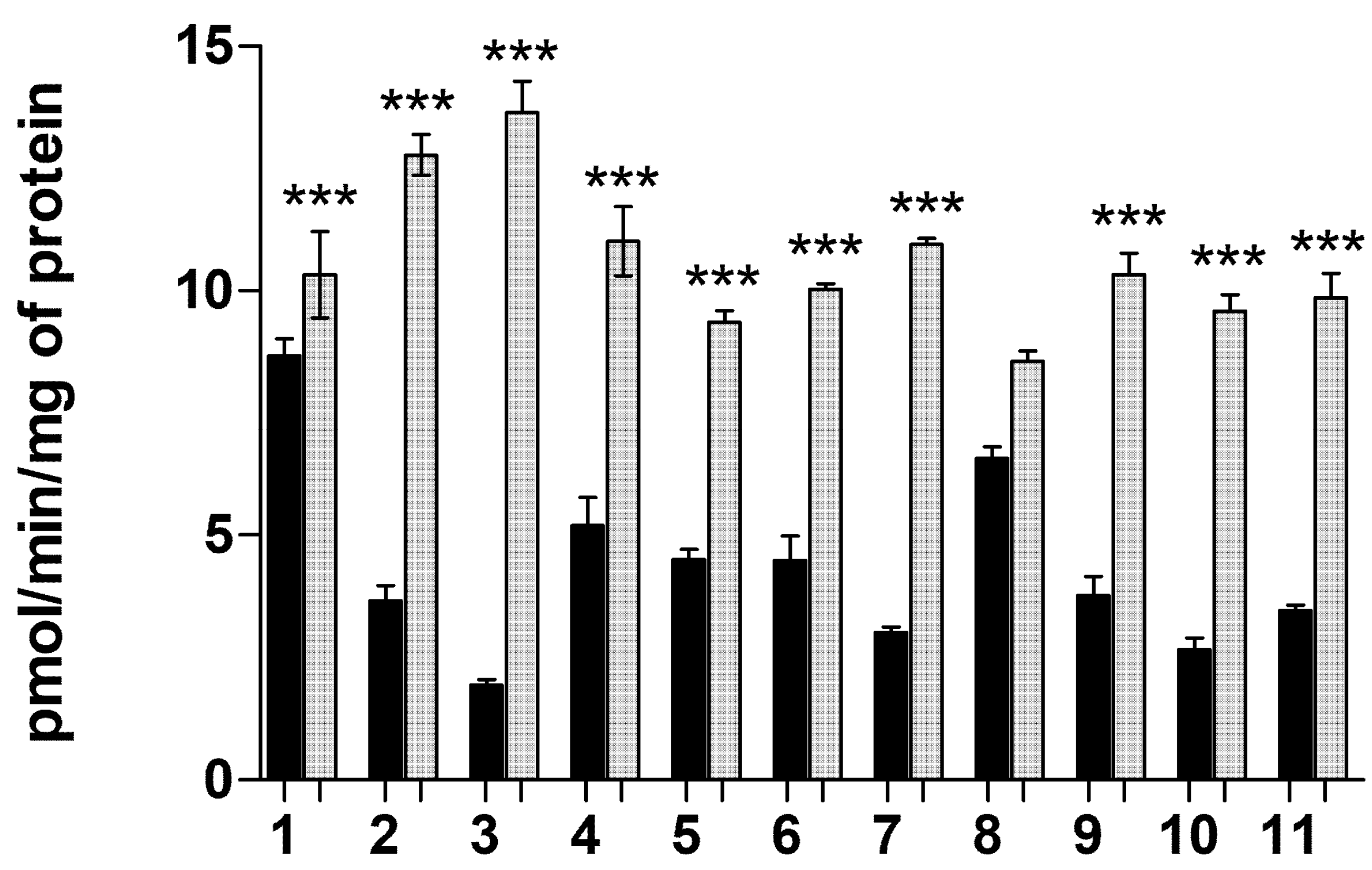
2.1.6. Transporter-Mediated Uptake of Neurosteroids 1–11 at pH 5.5
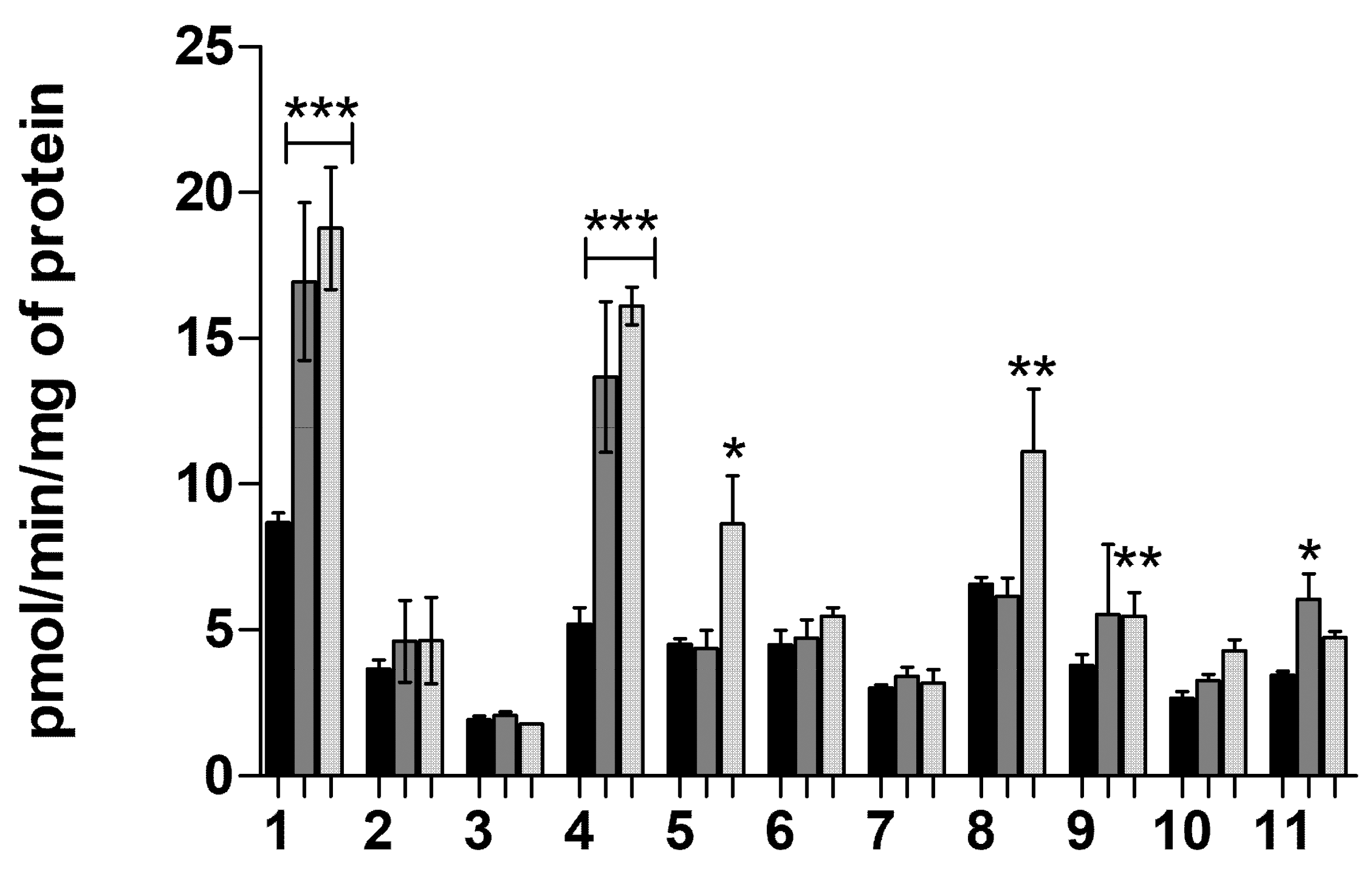
2.1.7. Transporter-Mediated Uptake of Neurosteroids 1–11 at pH 7.4
2.2. Molecular Modeling
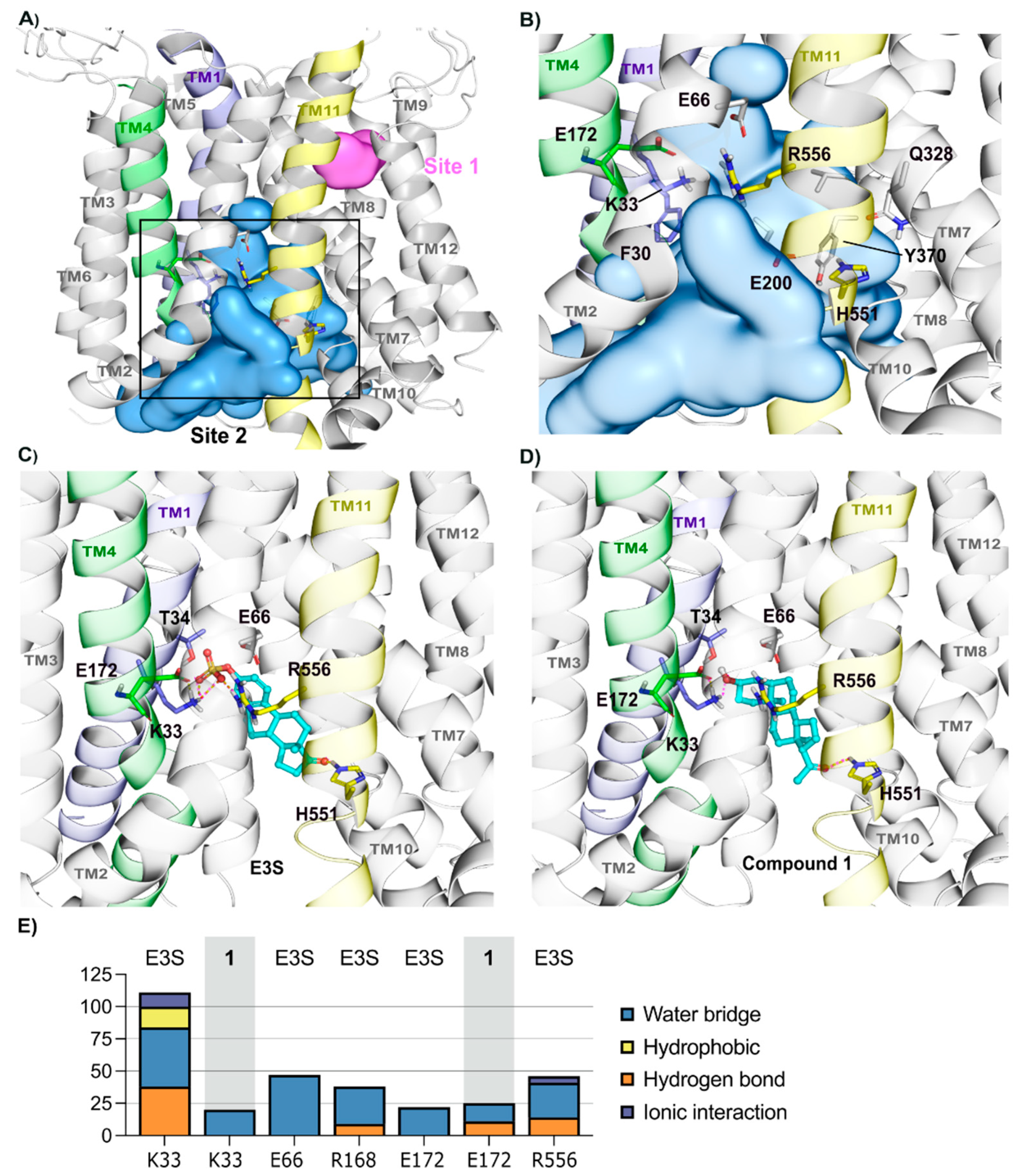
2.2.1. Homology Model—OATP1A2 Model Shows Two Binding Sites
2.2.2. OATP1A2 Substrate E3S Displays Two Potential Binding Modes
2.2.3. Binding of Compounds 2 and 3
2.2.4. Binding of Compounds 4 and 5
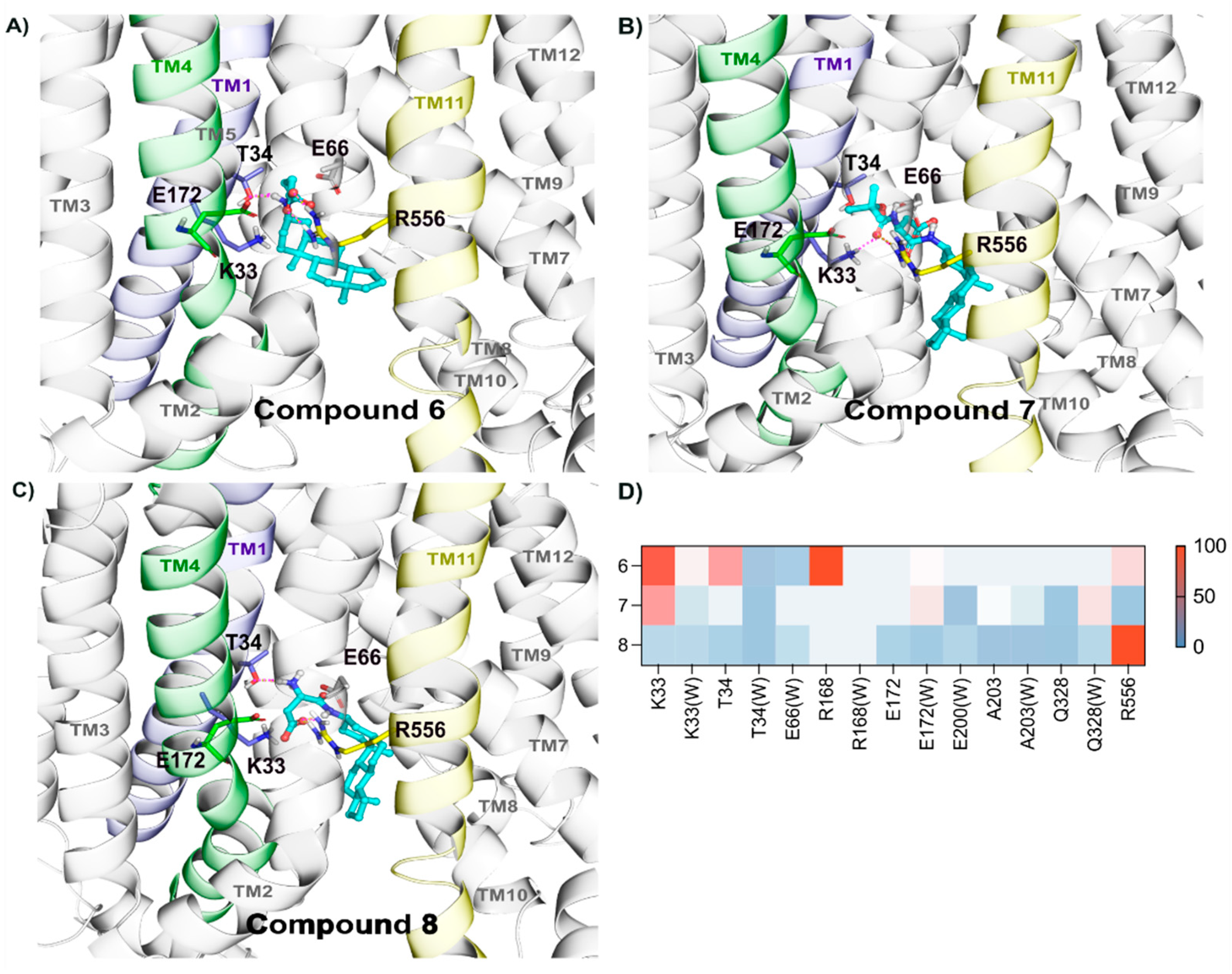
2.2.5. Binding of Compounds 6, 7, and 8
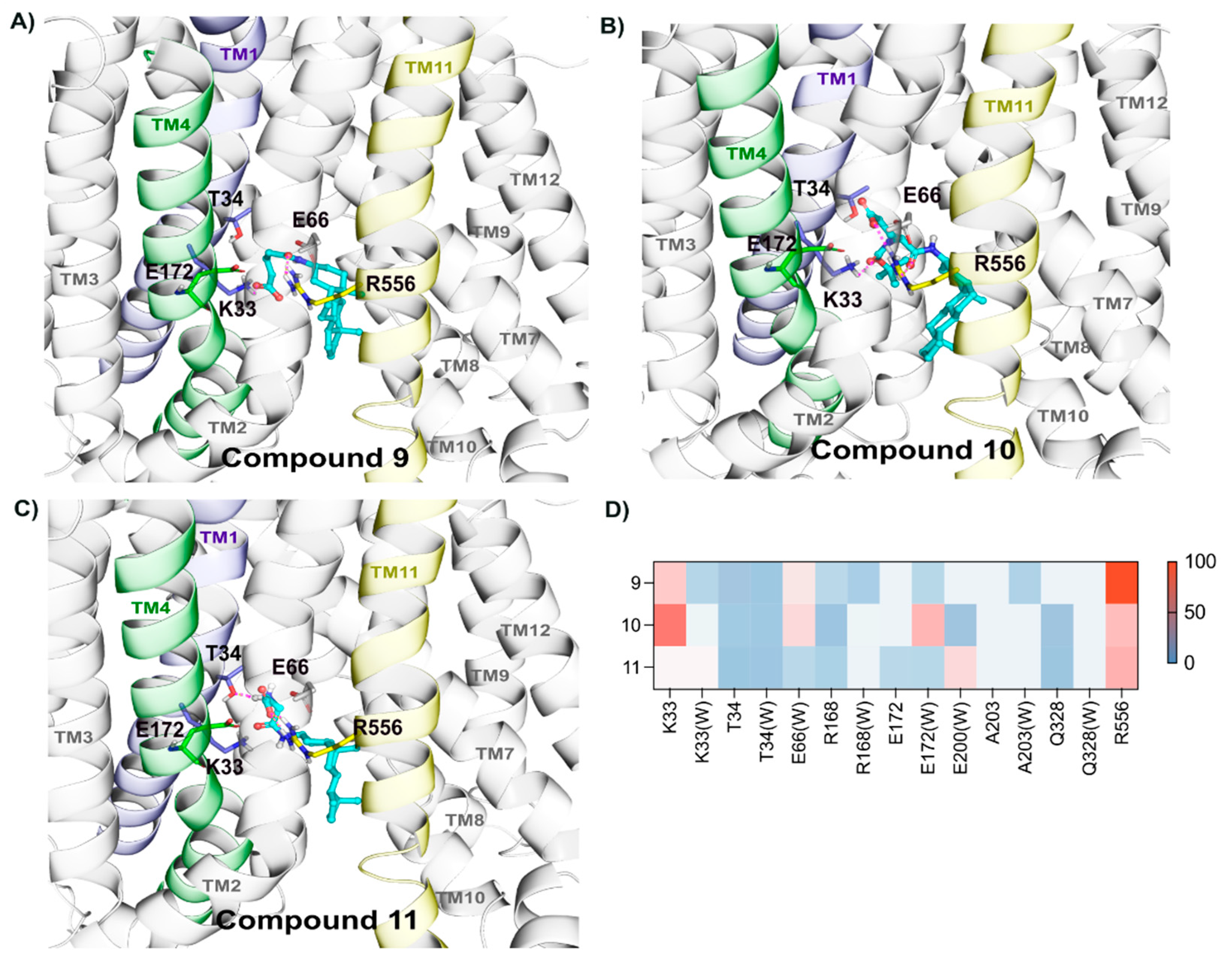
2.2.6. Binding of Compounds 9, 10, and 11
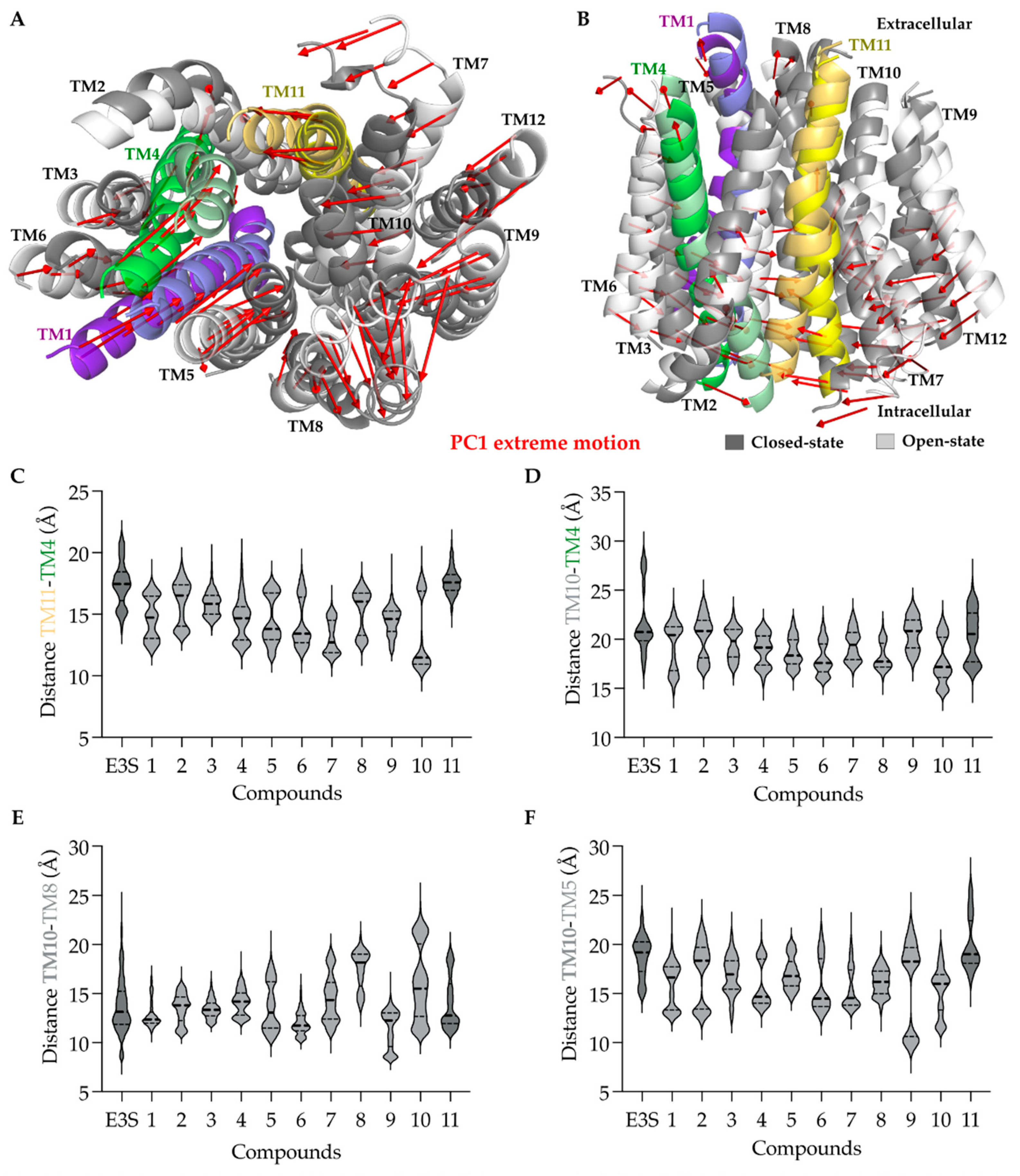
2.2.7. Principal Component Analysis (PCA)
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Cultures
4.3. Cellular Uptake of Neurosteroids
4.4. High-Performance Liquid Chromatography (HPLC) Analyses
4.5. Data Analysis
4.6. Molecular Modeling
4.6.1. Homology Model Generation and Protein Preparation
4.6.2. Binding Site Prediction
4.6.3. Ligand Preparation and Docking
4.6.4. Molecular Dynamics (MD) Simulations
4.6.5. Principal Component Analysis (PCA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shintani, E.Y. Donepezil: An anticholinesterase inhibitor for Alzheimer’s disease. Am. J. Health-Syst. Pharm. 1997, 54, 2805–2810. [Google Scholar] [CrossRef]
- Brewster, J.T.; Dell’Acqua, S.; Thach, D.Q.; Sessler, J.L. Classics in chemical neuroscience: Donepezil. ACS Chem. Neurosci. 2018, 10, 155–167. [Google Scholar] [CrossRef]
- Chen, Y.; Shohami, E.; Constantini, S.; Weinstock, M. Rivastigmine, a brain-selective acetylcholinesterase inhibitor, ameliorates cognitive and motor deficits induced by closed-head injury in the mouse. J. Neurotrauma 1998, 15, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.-G.; Han, L.-P.; Yang, Q.-Q.; Wang, M.-J.; Zhang, E.; Liu, H.-M. A practical and efficient stereoselective synthesis of (S)-rivastigmine and (R)-rivastigmine. ChemistrySelect 2018, 3, 1385–1387. [Google Scholar] [CrossRef]
- Pearson, V.E. Galantamine: A new Alzheimer drug with a past life. Ann. Pharmacother. 2001, 35, 1406–1413. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.H. Evaluation of memantine for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2003, 4, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Ebrahem, A.S.; Oremus, M. A pharmacoeconomic evaluation of cholinesterase inhibitors and memantine for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2018, 19, 1245–1259. [Google Scholar] [CrossRef]
- Lee, G.; Dallas, S.; Hong, M.; Bendayan, R. Drug transporters in the central nervous system: Brain barriers and brain parechyma considerations. Pharmacol. Rev. 2001, 53, 569–596. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Kell, D.B.; Oliver, S.G.; de Koning, H.P. How drugs get into cells: Tested and testable predictions to help discriminate between transporter-mediated uptake and lipoidal bilayer diffusion. Front. Pharmacol. 2014, 5, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, P.D.; Kell, D.B. Carrier-mediated cellular uptake of pharmaceutical drugs: An exception or the rule? Nat. Rev. Drug Discov. 2008, 7, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Girardin, F. Membrane transporter proteins: A challenge for CNS drug development. Dialogues Clin. Neurosci. 2006, 8, 311. [Google Scholar] [CrossRef]
- Kell, D.B.; Dobson, P.D.; Oliver, S.G. Pharmaceutical drug transport: The issues and the implications that it is essentially carrier-mediated only. Drug Discov. Today 2011, 16, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Davis, T.P. Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol. Rev. 2013, 65, 291–314. [Google Scholar] [CrossRef] [Green Version]
- Vallee, M.; Mayo, W.; Darnaudery, M.; Corpechot, C.; Young, J.; Koehl, M.; le Moal, M.; Baulieu, E.-E.; Robel, P.; Simon, H. Neurosteroids: Deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proc. Natl. Acad. Sci. USA 1997, 94, 14865–14870. [Google Scholar] [CrossRef] [Green Version]
- Mellon, S.H.; Griffin, L.D. Synthesis, regulation, and function of neurosteroids. Endocr. Res. 2002, 28, 463. [Google Scholar] [CrossRef]
- Morrow, A.L. Recent developments in the significance and therapeutic relevance of neuroactive steroids—Introduction to the special issue. Pharmacol. Ther. 2007, 116, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hosie, A.M.; Wilkins, M.E.; Smart, T.G. Neurosteroid binding sites on GABAA receptors. Pharmacol. Ther. 2007, 116, 7–19. [Google Scholar] [CrossRef]
- Wu, F.-S.; Chen, S.-C.; Tsai, J.-J. Competitive inhibition of the glycine-induced current by pregnenolone sulfate in cultured chick spinal cord neurons. Brain Res. 1997, 750, 318–320. [Google Scholar] [CrossRef]
- Yaghoubi, N.; Malayev, A.; Russek, S.J.; Gibbs, T.T.; Farb, D.H. Neurosteroid modulation of recombinant ionotropic glutamate receptors. Brain Res. 1998, 803, 153–160. [Google Scholar] [CrossRef]
- Sedláček, M.; Kořínek, M.; Petrovič, M.; Cais, O.; Adamusová, E.; Chodounská, H.; Ladislav, V., Jr. Neurosteroid modulation of ionotropic glutamate receptors and excitatory synaptic transmission. Physiol. Res. 2008, 57 (Suppl. 3), S49–S57. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Ferreira, I.L.; Rego, A.C. Dysfunctional synapse in Alzheimer’s disease—A focus on NMDA receptors. Neuropharmacology 2014, 76, 16–26. [Google Scholar] [CrossRef]
- Rotaru, D.C.; Yoshino, H.; Lewis, D.A.; Ermentrout, G.B.; Gonzalez-Burgos, G. Glutamate receptor subtypes mediating synaptic activation of prefrontal cortex neurons: Relevance for Schizophrenia. J. Neurosci. 2011, 31, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Ding, Q.; Chen, Z.; Yun, H.; Wang, H. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J. Biol. Chem. 2013, 288, 24151–24159. [Google Scholar] [CrossRef] [Green Version]
- Baulieu, E.E. Neurosteroids: A novel function of the brain. Psychoneuroendocrinology 1998, 23, 963–987. [Google Scholar] [CrossRef]
- Korinek, M.; Kapras, V.; Vyklicky, V.; Adamusova, E.; Borovska, J.; Vales, K.; Stuchlik, A.; Horak, M.; Chodounska, H.; Vyklicky, L. Neurosteroid modulation of N-methyl-D-aspartate receptors: Molecular mechanism and behavioral effects. Steroids 2011, 76, 1409–1418. [Google Scholar] [CrossRef]
- Karssen, A.M. Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology 2001, 142, 2686–2694. [Google Scholar] [CrossRef]
- Peng, R.; Zhang, H.; Zhang, Y.; Wei, D.-Y. Effects of the ABCB1 (1199G > A) polymorphism on steroid sex hormone-induced P-glycoprotein expression, ATPase activity, and hormone efflux. Med. Sci. 2015, 3, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, M.; Kusuhara, H.; Fujishima, M.; Adachi, Y.; Sugiyama, Y. Organic anion transporter 3 mediates the efflux transport of an amphipathic organic anion, dehydroepiandrosterone sulfate, across the blood-brain barrier in mice. Drug Metab. Dispos. 2011, 39, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Ducharme, N.; Banks, W.A.; Morley, J.E.; Robinson, S.M.; Niehoff, M.L.; Mattern, C. Brain distribution and behavioral effects of progesterone and pregnenolone after intranasal or intravenous administration. Eur. J. Pharmacol. 2010, 641, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Adla, S.K.; Slavikova, B.; Smidkova, M.; Tloustova, E.; Svoboda, M.; Vyklicky, V.; Krausova, B.; Hubalkova, P.; Nekardova, M.; Holubova, K.; et al. Physicochemical and biological properties of novel amide-based steroidal inhibitors of NMDA receptors. Steroids 2017, 117, 52–61. [Google Scholar] [CrossRef]
- Adla, S.K.; Slavikova, B.; Chodounska, H.; Vyklicky, V.; Ladislav, M.; Hubalkova, P.; Krausova, B.; Smejkalova, T.; Nekardova, M.; Smidkova, M.; et al. Strong inhibitory effect, low cytotoxicity and high plasma stability of steroidal inhibitors of N-methyl-D-aspartate receptors with C-3 amide structural motif. Front. Pharmacol. 2018, 9, 1299. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Huttunen, J.; Montaser, A.; Adla, S.K.; Auriola, S.; Lehtonen, M.; Huttunen, K.M. Ganciclovir and its hemocompatible more lipophilic derivative can enhance the apoptotic effects of methotrexate by inhibiting breast cancer resistance protein (BCRP). Int. J. Mol. Sci. 2021, 22, 7727. [Google Scholar] [CrossRef]
- Huttunen, J.; Gynther, M.; Vellonen, K.-S.; Huttunen, K.M. L-type amino acid transporter 1 (LAT1)-utilizing prodrugs are carrier-selective despite having low affinity for organic anion transporting polypeptides (OATPs). Int. J. Pharm. 2019, 571, 118714. [Google Scholar] [CrossRef] [PubMed]
- Hutzler, J.M.; Tracy, T.S. Atypical kinetic profiles in drug metabolism reactions. Drug Metab. Dispos. 2002, 30, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2). J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Yuan, J.; Li, Z.; Wang, Z.; Cheng, D.; Du, Y.; Li, W.; Kan, Q.; Zhang, W. Genetic polymorphisms and function of the organic anion-transporting polypeptide 1A2 and its clinical relevance in drug disposition. Pharmacology 2015, 95, 201–208. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Xu, S.; Ni, C.; Fang, Z.; Hong, M. Amino-terminal region of human organic anion transporting polypeptide 1B1 dictates transporter stability and substrate interaction. Toxicol. Appl. Pharmacol. 2019, 378, 114642. [Google Scholar] [CrossRef]
- Glaeser, H.; Mandery, K.; Sticht, H.; Fromm, M.; König, J. Relevance of conserved lysine and arginine residues in transmembrane helices for the transport activity of organic anion transporting polypeptide 1B3. Br. J. Pharmacol. 2010, 159, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, N.; Allen, C.; Bendayan, R. Differential role of organic anion-transporting polypeptides in estrone-3-sulphate uptake by breast epithelial cells and breast cancer cells. J. Pharmacol. Exp. Ther. 2012, 342, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kounnis, V.; Chondrogiannis, G.; Mantzaris, M.D.; Tzakos, A.G.; Fokas, D.; Papanikolaou, N.A.; Galani, V.; Sainis, I.; Briasoulis, E. Microcystin LR shows cytotoxic activity against pancreatic cancer cells expressing the membrane OATP1B1 and OATP1B3 transporters. Anticancer Res. 2015, 35, 5857–5865. [Google Scholar]
- Gong, I.Y.; Kim, R.B. Impact of genetic variation in OATP transporters to drug disposition and response. Drug Metab. Pharmacokinet. 2013, 28, 4–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-4: SiteMap; Schrödinger, LLC: New York, NY, USA, 2019.
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2019.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for PK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-4: MacroModel; Schrödinger, LLC: New York, NY, USA, 2019.
- Schrödinger Release 2019-4: Desmond Molecular Dynamics System; Maestro-Desmond Interoperability Tools; D.E. Shaw Research: New York, NY, USA; Schrödinger: New York, NY, USA, 2019.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transport Mechanism Type 1 | Transport Mechanism Type 2 | |||||
|---|---|---|---|---|---|---|
| Vmax (pmol/mg/min) | Km (µM) | Vmax/Km | Vmax (pmol/mg/min) | Km (µM) | Vmax/Km | |
| 1 | 26.3 ± 4.2 | 55.4 | 0.47 | - | - | - |
| 2 | n.d. a | n.d. a | n.d. a | |||
| 3 | 3.0 ± 0.4 | 7.7 | 0.39 | 8.2 ± 0.1 | 25.2 | 0.33 |
| 4 | 13.9 ± 6.7 | 232.8 | 0.059 | |||
| 5 | 5.3 ± 0.4 | 76.4 | 0.07 | 34.7 ± 7.7 | 959.6 | 0.04 |
| 6 | 3.9 ± 0.5 | 13.8 | 0.28 | 10.2 ± 0.1 | 91.26 | 0.11 |
| 7 | 3.0 ± 0.1 | 3.0 | 1.0 | 23.0 ± 0.5 | 127.4 | 0.18 |
| 8 | 8.0 ± 2.1 | 10.9 | 0.73 | 28.2 ± 0.1 | 42.8 | 0.66 |
| 9 | 5.5 ± 0.6 | 9.2 | 0.60 | 26.5 ± 3.0 | 67.8 | 0.39 |
| 10 | 15.4 ± 6.5 | 23.0 | 0.67 | 150.4 ± 9.9 | 281.6 | 0.53 |
| 11 | 17.2 ± 2.1 | 24.2 | 0.71 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adla, S.K.; Tonduru, A.K.; Kronenberger, T.; Kudova, E.; Poso, A.; Huttunen, K.M. Neurosteroids: Structure-Uptake Relationships and Computational Modeling of Organic Anion Transporting Polypeptides (OATP)1A2. Molecules 2021, 26, 5662. https://doi.org/10.3390/molecules26185662
Adla SK, Tonduru AK, Kronenberger T, Kudova E, Poso A, Huttunen KM. Neurosteroids: Structure-Uptake Relationships and Computational Modeling of Organic Anion Transporting Polypeptides (OATP)1A2. Molecules. 2021; 26(18):5662. https://doi.org/10.3390/molecules26185662
Chicago/Turabian StyleAdla, Santosh Kumar, Arun Kumar Tonduru, Thales Kronenberger, Eva Kudova, Antti Poso, and Kristiina M. Huttunen. 2021. "Neurosteroids: Structure-Uptake Relationships and Computational Modeling of Organic Anion Transporting Polypeptides (OATP)1A2" Molecules 26, no. 18: 5662. https://doi.org/10.3390/molecules26185662
APA StyleAdla, S. K., Tonduru, A. K., Kronenberger, T., Kudova, E., Poso, A., & Huttunen, K. M. (2021). Neurosteroids: Structure-Uptake Relationships and Computational Modeling of Organic Anion Transporting Polypeptides (OATP)1A2. Molecules, 26(18), 5662. https://doi.org/10.3390/molecules26185662