3.1. Carbohydrate or Sugar-Based Inhibitors
Some inhibitors belonging in this huge family include substances that bind to the active sites of STs in competition with natural substrates but cannot undergo sialylation because they lack the required acceptor groups. These compounds have been developed for several mammalian and bacterial STs, which exhibit strict specificity for substrates containing terminal
N-acetyllactosamine or lactose moieties at the non-reducing ends of glycoconjugates [
104,
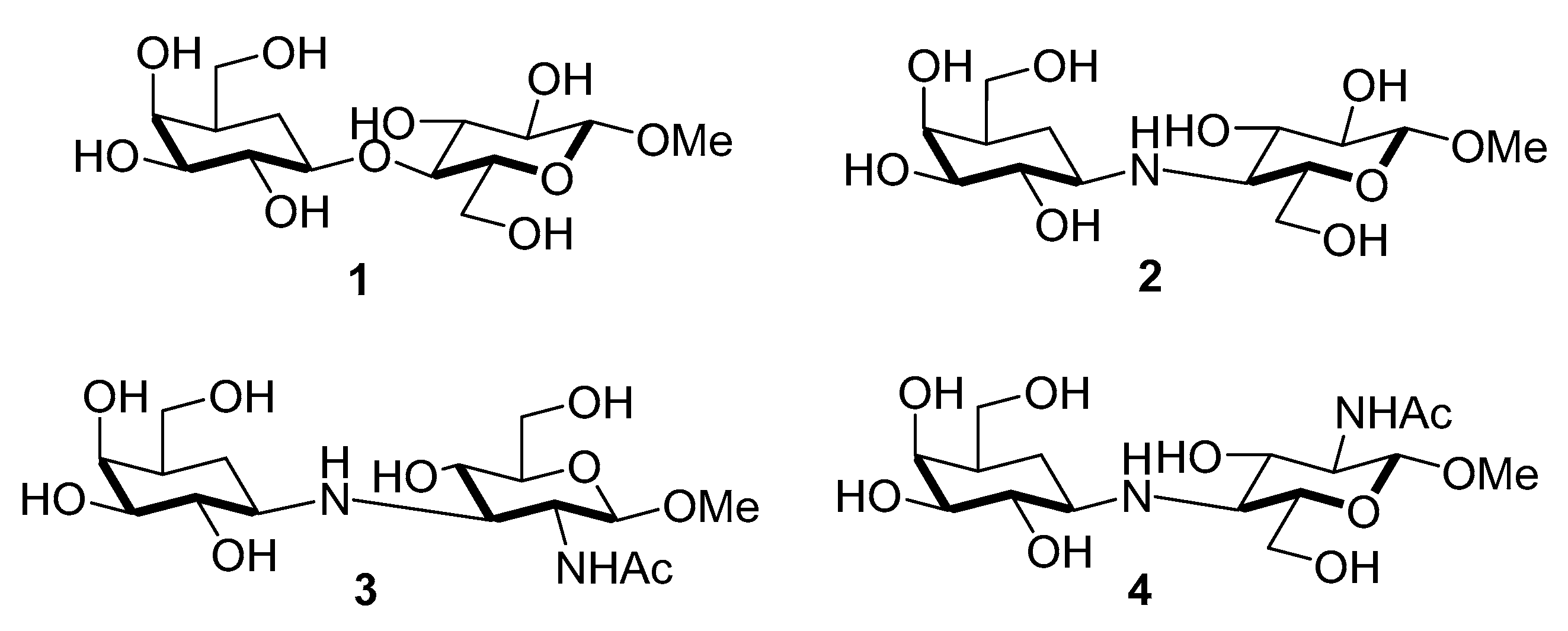
105]. Ogawa and coworkers found that the methyl 5a’-carbadisaccharides
1–
4 (
Figure 3), in which the monosaccharide units are linked via ether or amine bridges, possessed considerable inhibitory activities (IC
50 = 185–419 μM) towards recombinant α2,3-sialyltransferases using 4-methylumbellipheryl-labeled LacNAc as the acceptor substrate (
Table 2) [
106]. The inhibitory activities of
1–
4 towards rat liver α2,6-sialyltransferases were also determined and shown in
Table 2.
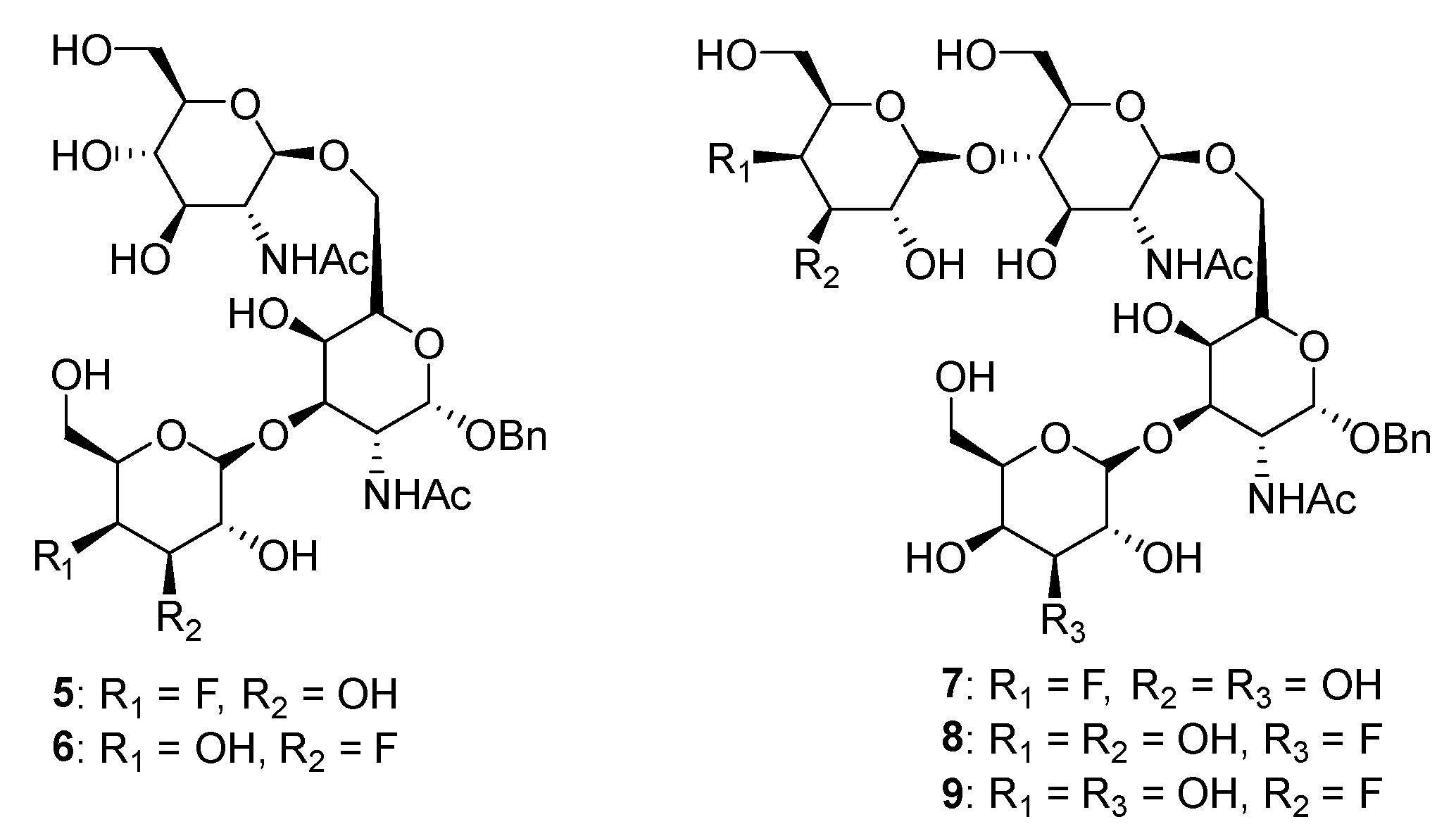
Matta’s group designed and synthesized several fluorinated mucin core 2 branched oligosaccharides (
5–
9,
Figure 4) [
107]. The results demonstrated that compound
7 had a modest inhibitory activity (inhibition constant, K
i = 1.9 mM) against cloned α2,6-(N)-ST (rST6Gal-I) but not α2,3-(N)-ST (rST3Gal-III). Interestingly, other substances in this group did not display inhibitory properties against STs. Moreover,
8 and
9 served as good acceptor substrates of α2,6-(N)-ST/α2,3-(N)-ST and cloned α2,3-(O)-ST/prostate cancer cell LNCaP α2,3-(O)-ST, respectively. Their findings indicated that the position of fluorine substitution on the mucin core 2 branched oligosaccharides affects the nature of the carbohydrates-enzyme interactions and, as a result, determines whether these substances can serve as ST substrates (acceptor) or inhibitors. To this date, no recent acceptor analogue ST inhibitors were reported.
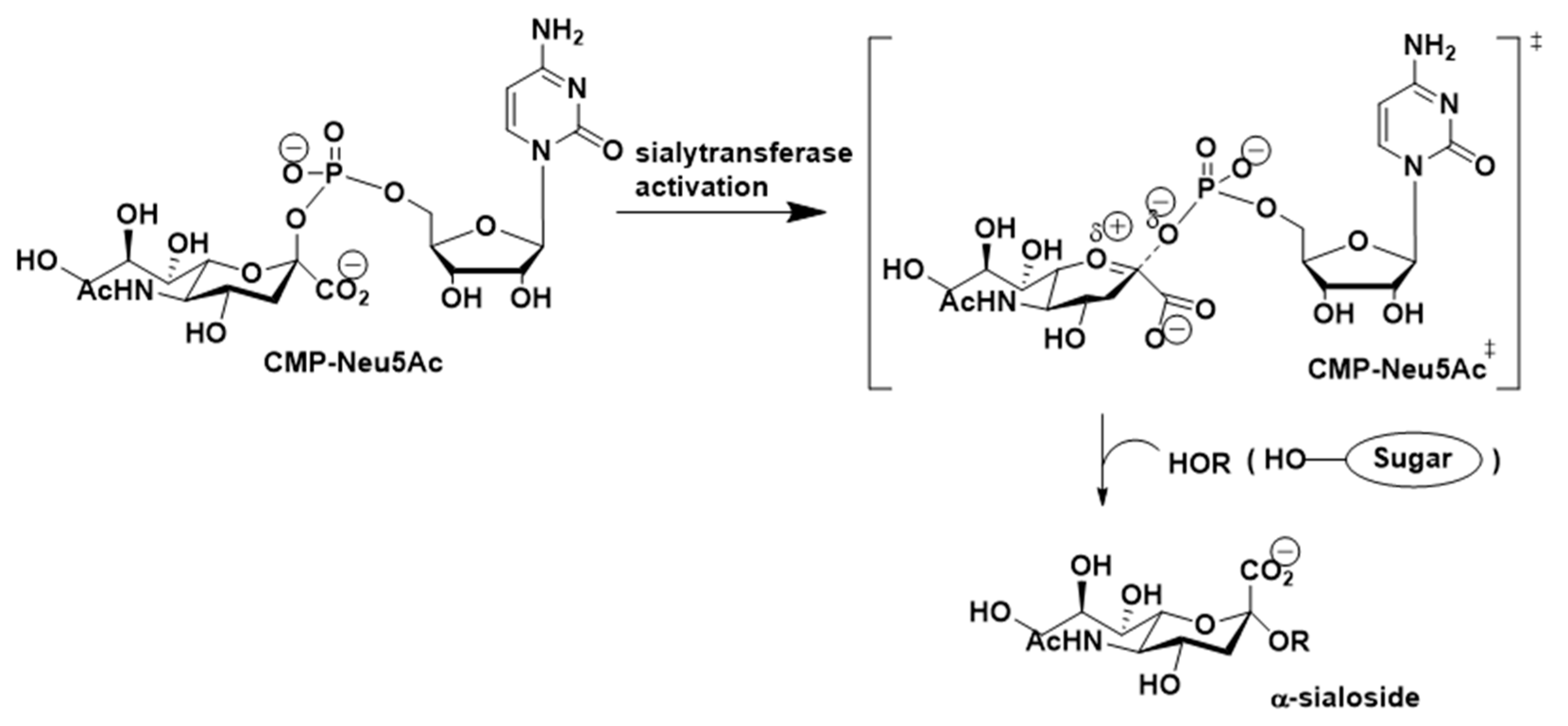
Several studies conducted by Horenstein and Schmidt aimed at gaining insights into the detailed mechanism of the ST-promoted sialylation reactions. The observations made through this effort suggested that sialyltransferase-catalyzed reactions of the sialyl donor, (CMP-Neu5Ac), proceed through a transition state in which the leaving group (CMP) departs before bond formation with the incoming hydroxyl nucleophile. The transition state (
Figure 5) for this process is believed to display oxocarbenium ion-like character with the carbohydrate ring existing in a distorted half-chair conformation and possessing a considerable amount of positive charge on the anomeric carbon atom [
108,
109,
110,
111,
112,
113].
Building upon this idea, several transition state analogues have been designed and tested as ST inhibitors. Most of these substances—which contain (i) a planar anomeric carbon, (ii) an increased distance between the CMP leaving group and the anomeric carbon, and (iii) at least two negative charges close to the site that mimics glycosylation cleavage—exhibited high affinities to sialyltransferases [
113,
114,
115,
116,
117,
118,
119,
120].
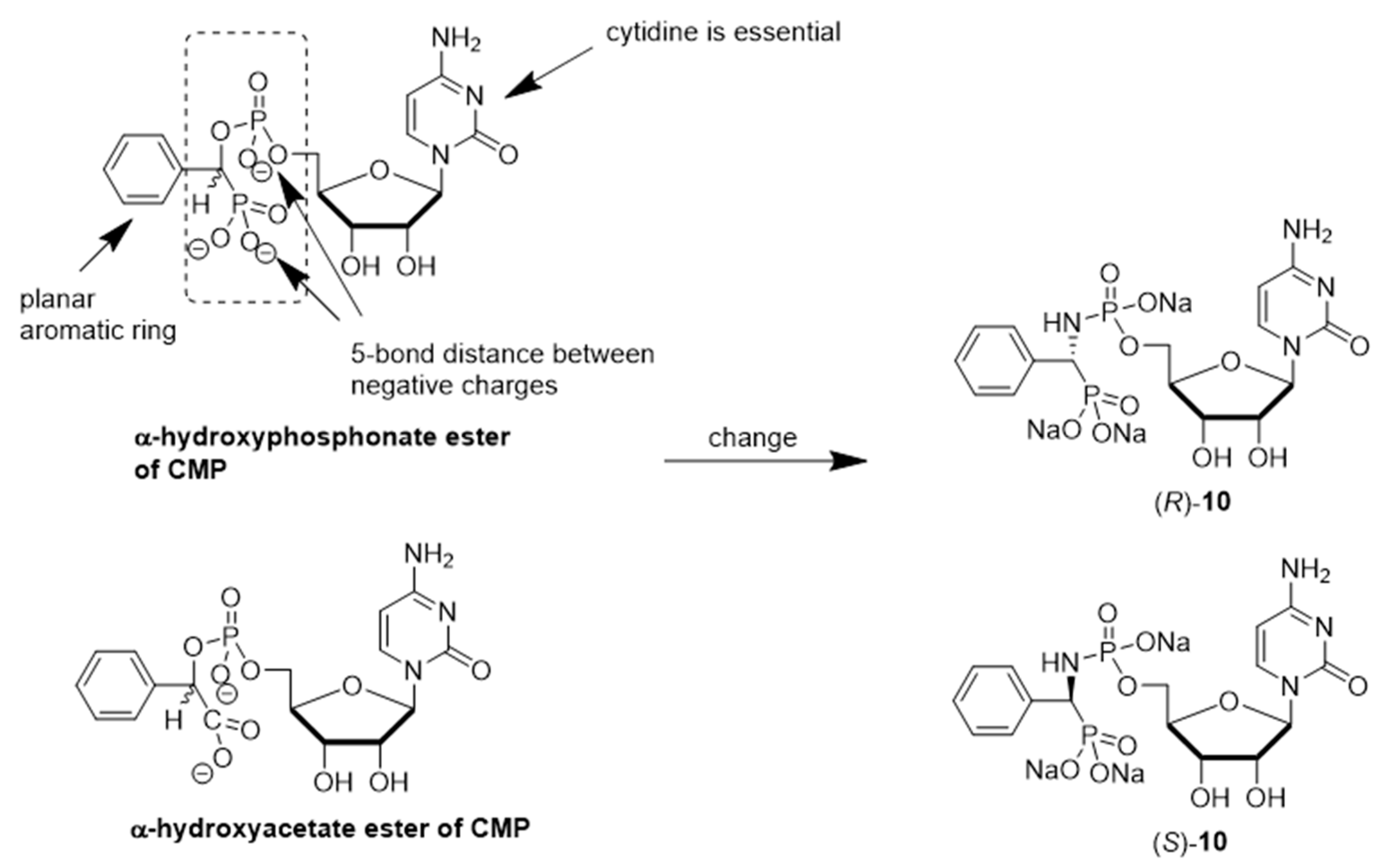
Schmidt and coworkers prepared the enantiomeric phosphoramidate-linked substances, (
R)-10 and (
S)-10 (
Figure 6), which possess
N-(α-phosphoryl)-phosphoramidate nucleosides and aromatic moieties, as potential ST inhibitors. These phosphoramidates mimic the CMP-Neu5Ac portion of the transition state structure in the sialylation reaction [
121]. The results of this study showed that (
R)-10 and (
S)-10 are both competitive inhibitors of α2,6-ST with respective K
i values of 68 ± 24 and 140 ± 30 μM. Moreover, the presence of
N-(α-phosphoryl)-phosphoramidate nucleoside and aromatic moieties in (
R)-10 resulted in a binding affinity towards rST6Gal-I that is comparable to that of the neutral substrate CMP-Neu5Ac (K
m = 46 ± 7 μM).
Cytidine diphosphate (CDP), a potent competitive inhibitor (K
i = 10 μM) of sialyltransferases, mimics the CMP portion of the donor substrate CMP-Neu5Ac. Despite its potency, CDP and its analogues have not been subjected to thorough biological studies as inhibitors of STs [
114]. On the other hand, the Fukuda group synthesized donor analogues 5-methyl CMP and 2′-O-methyl CMP as polysialyltransferase inhibitors. Their results revealed that 2′-O-methyl CMP strongly inhibited ST8Sia-IV, ST8-Sia-II, and ST8Sia-III. Upon treatment with the inhibitors, polysialic acid expression on the cell surface was reduced [
97,
122]. Our efforts identified a group of 5′-triazole nucleosides that are analogues of CMP using a substrate-based drug design approach. These substances, containing a unique 1,2,3-triazole subunit, were synthesized using Cu(I)-catalyzed Huisgen 1,3-cycloaddition reactions (click chemistry) [
123,
124]. Among the CMP analogues tested, most exhibited low inhibition of rST3Gal-I (
Table 3). Only compound
11 was found to be more potent with an IC
50 of 37.5 μM which suggests that the aromatic functionality and the cytosine group are required for better binding in the enzyme active site.
To gain a more thorough understanding of how binding of sugar-based ST inhibitors is governed by molecular shape, charge, H-bonding, and hydrophobic interactions, Zou and coworkers designed three types of substances including substrate mimics containing the 2-deoxy-2,3-dehydro-acetylneuraminic moiety and derivatives of aryl sulfonamides attached to cytidine [
125]. One subgroup of these substances including
13–
14 (
Table 4) are substrate mimics in that they possess a non-hydrolysable and uncharged 1,2,3-triazole moiety as the linker rather than a phosphate group [
126,
127]. The second group of ST inhibitors includes
15–
21, all of which has a 2-deoxy-2,3-dehydro-acetylneuraminic moiety attached to cytidine through different carboxylic acid and amide linker groups [
128]. The third type of substances represented by
22–
39 possess an aryl sulfonamide group that replaced the sialylphosphate group in the CMP-Neu5Ac structure. All these designs are driven by the fact that the charged phosphate linker provides poor cellular permeability and instability towards phosphatases. Among these substances,
35 was observed to have the highest inhibitory activity against Campylobacter jejuni sialyltransferase (CJ ST) Cst 06 with a K
i value of 87 μM [
129]. Other members of this series except for
13,
14,
34, and
37 displayed low activities. It is however noteworthy that the first group of compounds are competitive inhibitors of CJ ST, while members of the other two subfamilies inhibit in a non-competitive fashion. These observations suggest that the charge and hydrophobic character, rather than molecular shape and H-bonding interactions, contribute more to the tight binding of these compounds against CJ ST. Therefore, it seems that a more practical way to improve the drug-like properties of these inhibitors is not to totally eliminate the charge but to mask it temporarily as a pro-drug.
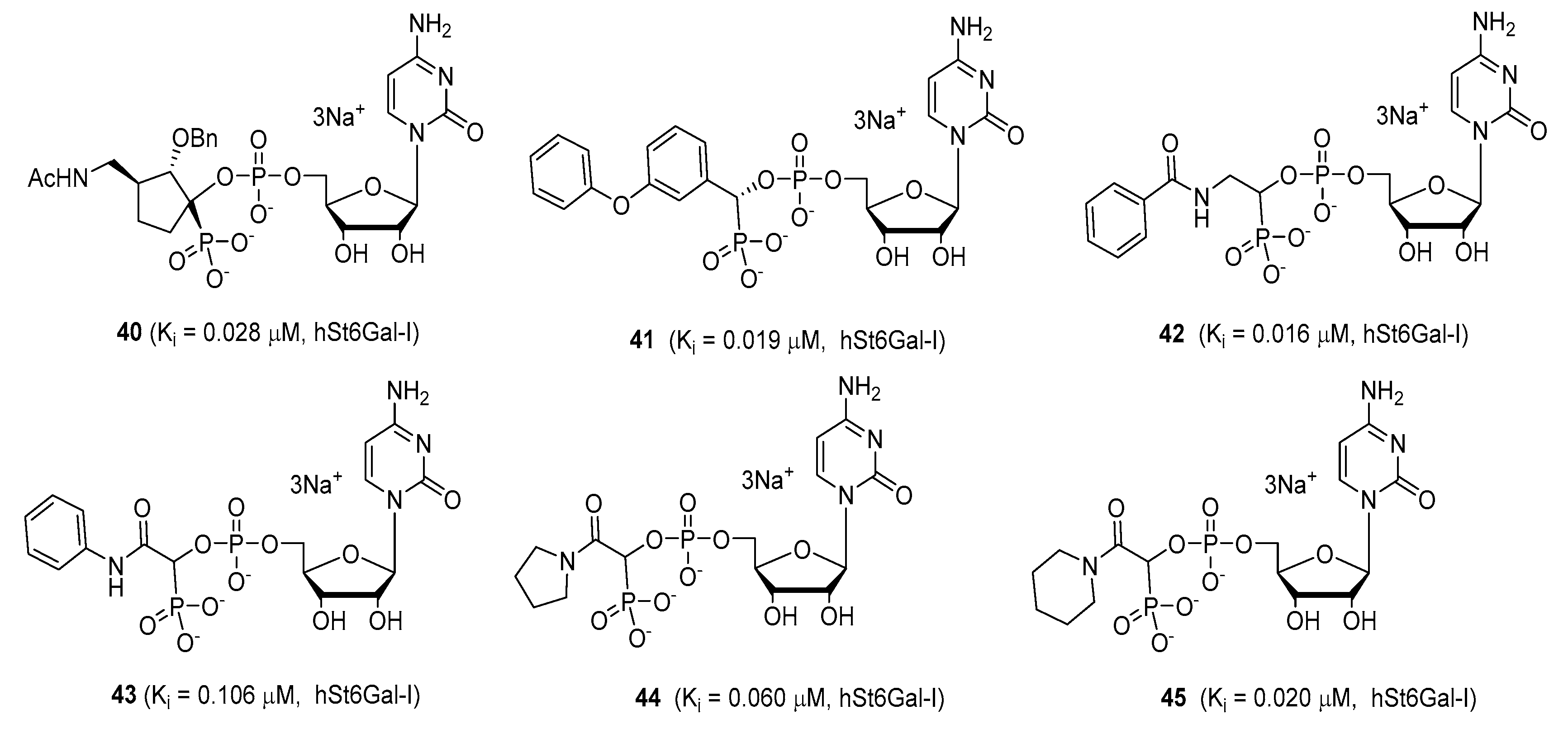
Another set of cyclopentane-based transition state analogues were designed and prepared by Ye and others [
130]. Their design is based on the idea that the cyclopentane skeleton in its puckered conformation may mimic the planar structure of the donor in the transition state. Hence, in this study, the Neu5Ac moiety in the donor was replaced by cyclopentyl α-hydroxyphosphonates and the substituent effects on the five-membered ring were analyzed. A potent inhibitor
40 with a K
i of 0.028 ± 0.006 μM was identified (
Figure 7), slightly weaker than the previously reported 3-phenoxybenzyl-based inhibitor [
119] from Schmidt’s group
41 which has a K
i of 0.019 ± 0.0065 μM [
130]. A more recent work by the same group explored the attachment of an amide group to CMP to mimic the geometry and charge distribution in the transition state. This new set of compounds
42–
45 possessed excellent α2,6-sialyltransferase inhibitory activities (
Figure 7), which suggests that the amide group can be a facile and interesting structural isostere for transition state ST inhibitors [
131].
Even though the most potent ST inhibitors reported are these transition state analogues, such compounds suffer from low lipophilicity and poor pharmacokinetic properties and are generally synthetically challenging. Skropeta and others took advantage of the availability of the structure of human ST8Sia-III to perform a structure-based computer-aided design [
132]. Computer simulations revealed that among the transition state-based inhibitors studied, there was no significant difference in their calculated binding affinities upon substitution of cytidine with a uridine moiety as well as between R and S diastereomers. Molecular dynamics simulations of proposed carbamate- and triazole-linked analogues showed similar binding to the enzyme’s active site relative to other reported analogues. Such proposed neutral linkers are attractive for they may address the poor pharmacokinetic properties brought about by the phosphodiester linkage in previously developed ST inhibitors and may even offer potential selectivity towards hST8Sia-III [
132]. The same group performed a similar study using the crystal structure of human ST6Gal-I [
133]. In this work, it was demonstrated that compounds with a neutral carbamate or triazole linker maintained key interactions in the active site of the enzyme. Interestingly, the neutral linkers were slightly more favorable than the charged phosphodiester moiety, suggesting that a carbamate or 1,2,3-triazole group may serve as a bioisostere of the phosphodiester group in the development of novel inhibitors. Thus, the same group recently published the synthesis and ST6Gal-I inhibition studies of 24 carbamate-linked uridyl-based compounds [
134]. Among them, five promising compounds (
46–
50) (
Table 5) exhibited K
i’s in the range of 1–20 μM. Given the potency of these ST inhibitors in vitro, it would be very interesting to understand their mechanism of action and investigate their functional effects in vivo.
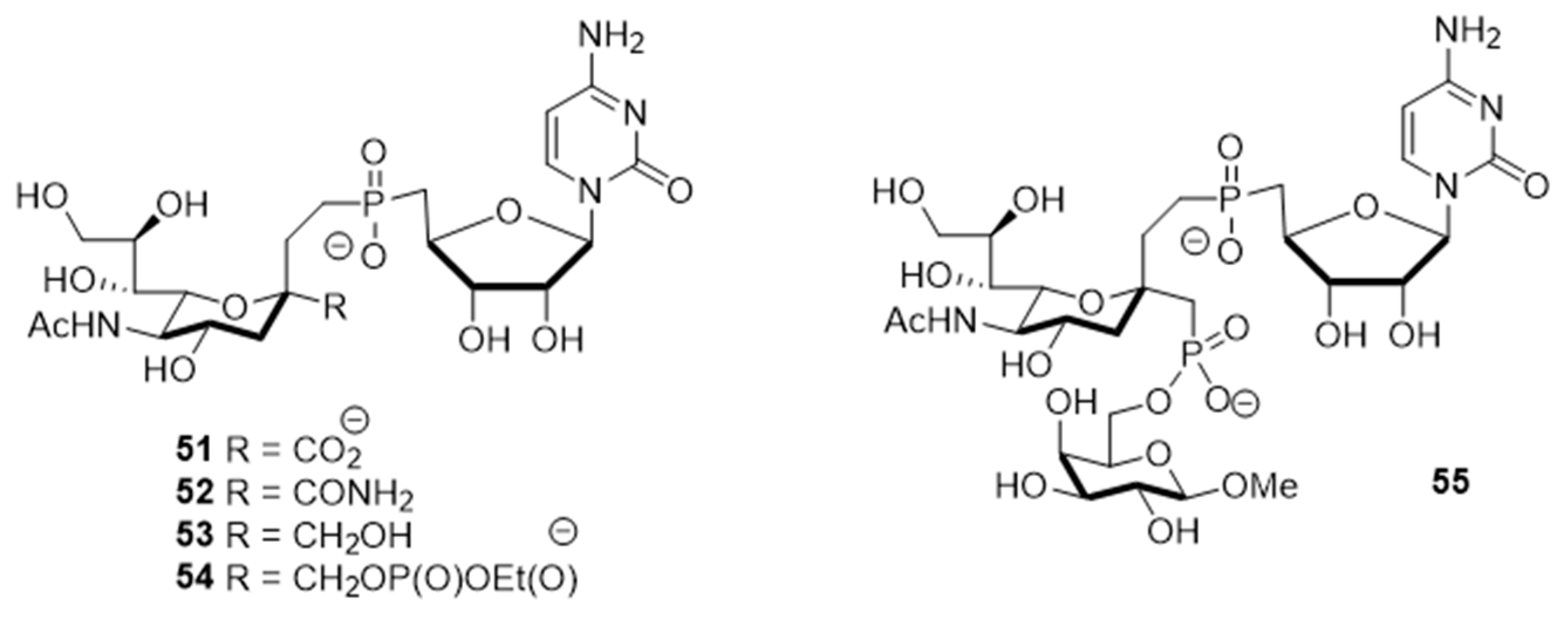
The utility of bisubstrate-type substances, such as those that contain both donor (CMP-Neu5Ac) and acceptor (
N-acetyllactosamine, LacNAc) components, as sialyltransferase inhibitors was already recognized earlier [
135]. Izumi and coworkers designed the bisubstrate analogue
55, containing the donor-like substrate (CMP-Neu5Ac mimic) and the acceptor substrate (galactose) [
136]. Four donor analogues,
51–
54, possessing the partial structure of the bisubstrate analogue, were used as controls in this structure-activity relationship study. The unique structural feature in each analogue is an ethylene group that replaced the exocyclic anomeric oxygen of CMP-Neu5Ac. Donor analogues
51–
54, possessing different replacements for the C-1 carboxylate group of the Neu5Ac moiety (carboxyamide, hydroxymethyl, or methylene phosphate), displayed decreased inhibitory activities against rat recombinant α2,3- and α2,6-ST. Among these substances, cytidin-5′-yl sialylethylphosphonate
51 gave moderate inhibitory activities with respective IC
50 values of 0.047 and 0.34 mM against two rat recombinant α2,3- and α2,6-ST, which are 10−100-fold lower than those of
52 and
53 (
Figure 8 and
Table 6). Unlike
51, the bisubstrate analogue
55 has less potent inhibitory activities against rat recombinant α2,3- and α2,6-ST (IC
50 = 1.3 and 2.4 mM, respectively). As evident from inspection of the data displayed in
Table 6, insertion of a charged group and extension of the C-1 carboxylate did not lead to the enhancement of inhibitory potency towards STs. Therefore, refinement of this strategy for designing bisubstrate inhibitors remains necessary.
As depicted in
Figure 5, transfer of sialic acids to terminal positions of oligosaccharide chains of glycoconjugates catalyzed by ST is believed to proceed through a mechanism involving an oxocarbenium ion-like transition state. This mechanistic proposal suggests that incorporation of fluorine on the pyranose ring of sialic acid would give fluorinated CMP-sialic acids that may be weak ST substrates but perhaps good ST inhibitors [
137,
138]. This proposal is based on the likelihood that strongly electronegative substituents such as F, incorporated especially at C-3 position of the ring, would destabilize the forming cationic transition state and result in a decrease in substrate activity without affecting binding to STs. The results of studies validated this proposal by showing that commercially available CMP-3FNeu5Ac inhibited recombinant α2,6-ST and was utilized as a chemical probe for mechanistic, kinetic, and structural studies of STs and related enzymes such as sialidases [
139,
140].
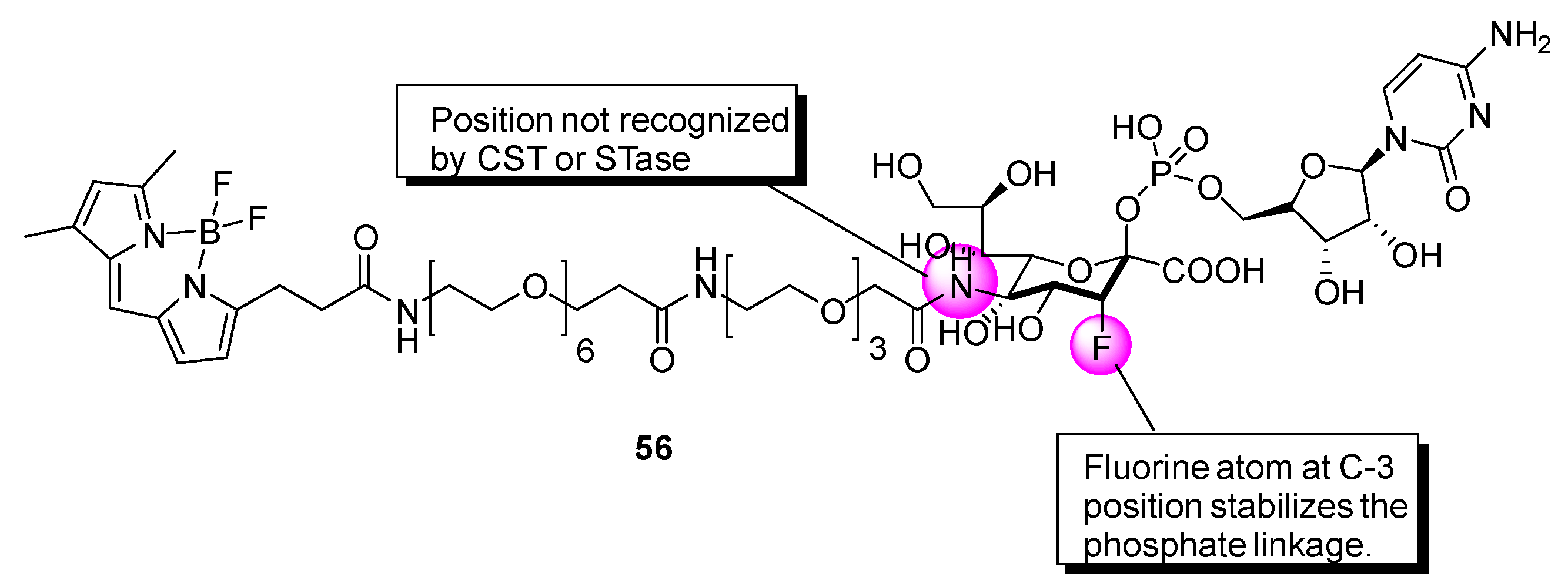
Further investigations have been carried out to expand the mechanistic understanding of the CMP-sialic acid transporter (CST)-promoted transport of CMP-sialic acid from the cytoplasm into the lumen of the Golgi apparatus [
141]. Kanie and coworkers conducted studies with the fluorescent CMP-3′′-F-Sia derivative
56 (
Figure 9), which is composed of a CMP-β-3′′-F-Sia moiety linked to a fluorescent tag [
142]. This probe was found to have an inhibitory potency (K
i = 31.7 μM) against rST3Gal-III relative to that of the parent compound lacking the fluorophore (K
i = 5.7 μM, [rST6Gal-I]) [
139,
143,
144]. Uptake of
56 into isolated Golgi vesicles of rat liver was determined using a competitive import test with CMP-Sia, which retarded the accumulation of
56, suggesting that
56 can be employed as a substrate for CST in studies designed to gain information about glycan processing events.
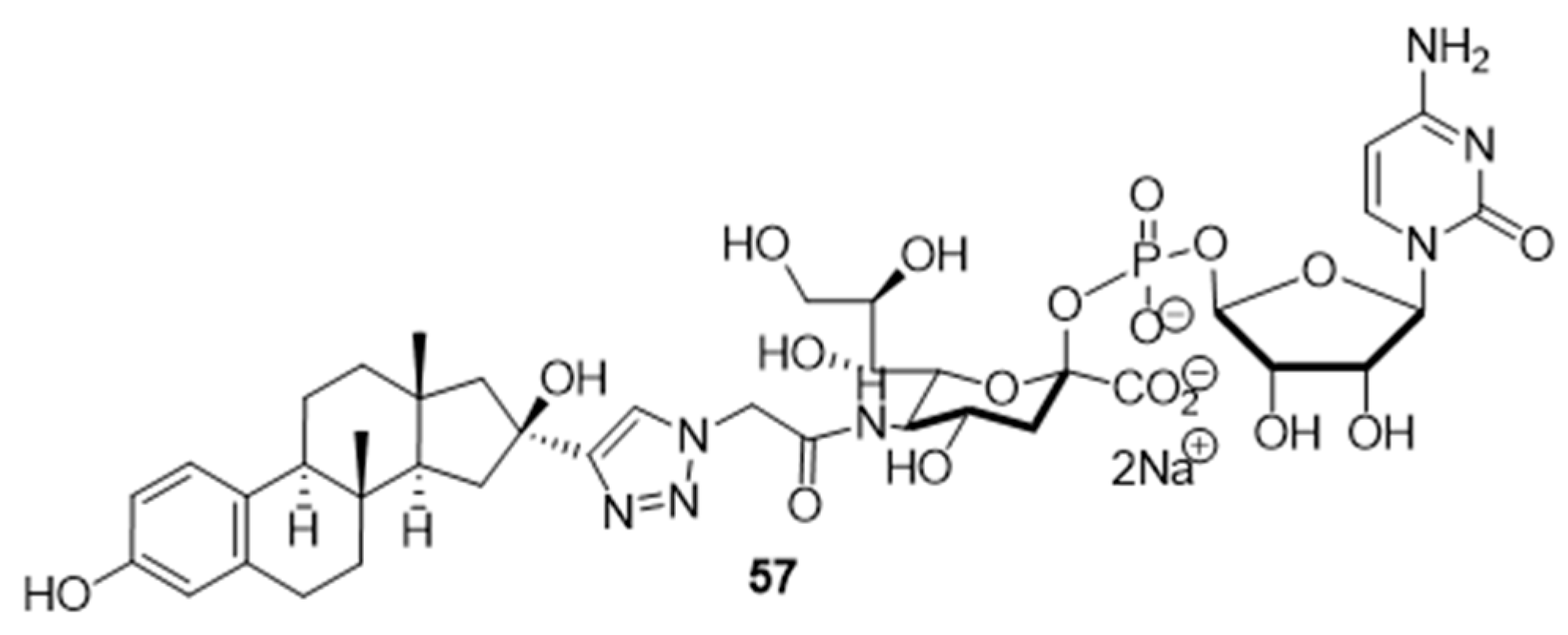
At the current time, efforts are focused on the rapid identification of potent protein inhibitors (including ST inhibitors) using high-throughput screening (HTS) approaches. In this regard, the MS-based, rapid, and quantitative screening technique developed by Nishimura and coworkers is particularly attractive [
145]. In this method, the inhibitory effects of substances on STs were determined quantitatively in the presence of CMP-Neu5Ac and an internal standard acceptor labelled with a stable isotope (OCD
3). The assay was carried out by comparing the intensities of MS peaks of the expected products possessing OCH
3 or OCD
3 moieties in the absence and presence of inhibitors. A focused library of non-natural sugar triazole nucleotides was constructed by using click chemistry to link azidosugar nucleotides and various alkynes. Among the sugar triazole nucleotides prepared in this manner,
57, possessing a steroid moiety, intriguingly displayed the highest inhibitory activity with an IC
50 value of 8.2 μM against rST3Gal-III (
Figure 10). It is very interesting that
57 was also accepted as a substrate (K
m = 125 μM) of rST6Gal-I [
145]. It is most likely that multiple mechanisms are operating in the substrate and inhibition functions of the corresponding STs in living cells. Owing to this observation, the mechanism of
57 to suppress ST activity inside the cell remain unexplored. It is also worth mentioning that even very bulky substitutions (i.e., steroids) at the 5- or 9-position of the sialic acid portion did not hinder the sialylation reaction.
The difficulty of developing membrane permeable donor nucleotide analogues has been demonstrated to be a consequence of their highly hydrophilic and negatively charged nature. However, previous studies have shown that glycosyltransferase-mediated donor nucleotide biosynthetic pathways in cells tolerate a range of sugar analogues whether of natural or unnatural origin [
146,
147]. These results suggest the potential of a new strategy for the design of glycosyltransferase inhibitors that are active in cells which focuses on promiscuous monosaccharide salvage pathways and metabolic feedback loops to globally shutdown the function of STs.
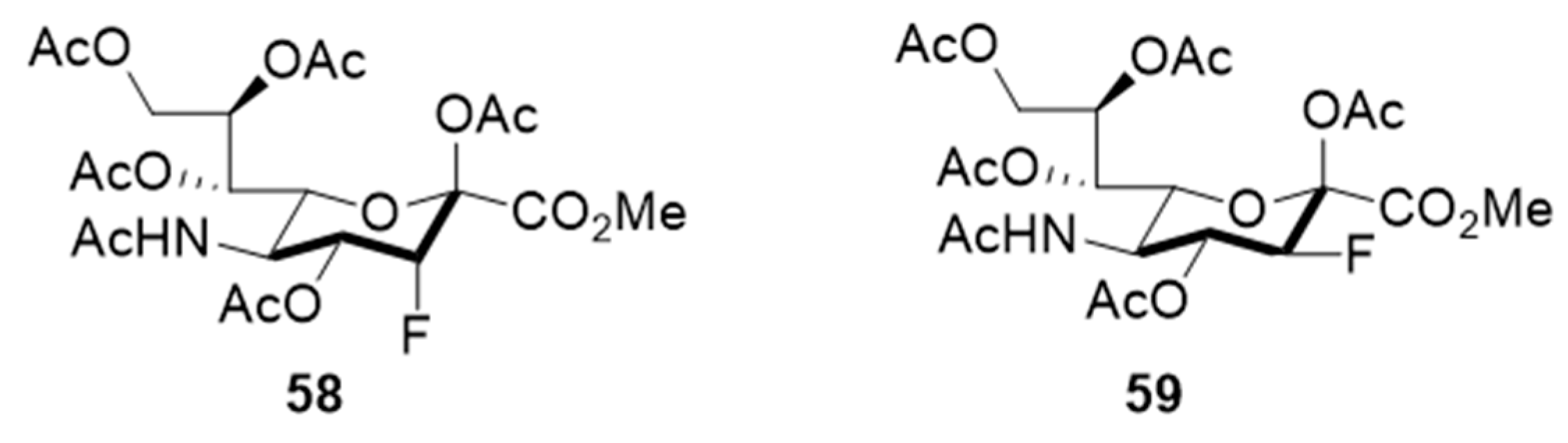
In line with the observed relaxed substrate specificity of STs in sialic acid salvage pathways, the results of an investigation by Paulson and coworkers demonstrated that the global metabolic inhibitors of STs remodeled the glycome [
137] because they are not used as a substrate but rather are bound to the active site of the enzyme. Specifically, it was found that peracetylated sialic acids
58–
59, bearing a fluorine at the position proximal to the endocyclic oxygen of the sugar moiety, were taken up and intracellularly metabolized to form the corresponding nucleotide sugar donor analogues (
Figure 11). This activity effectively collapsed the synthesis of a range of sialylated glycan epitopes and altered the cellular glycome. The fact that membrane permeable fluorinated analogues of sialic acid are inhibitors of STs and have the potential to regulate glycosylation in vitro, might pave the way for the use of these metabolic inhibitors to explore the roles of sialylated glycans in cancers. As a matter of fact, compound
58 (3F
ax-Neu5Ac) depleted α2,3- and α2,6-linked sialic acids in B16F10 cells, diminished migratory capacity, and reduced in vivo tumor growth [
138]. As far as we know, this is the first in vivo investigation for sugar-based ST inhibitors and should pave way to further developments. The most recent report revealed that ST3Gal-VI inhibition using the pan-ST inhibitor
58 (3F
ax-Neu5Ac) resulted in improved in vivo survival by increasing drug sensitivity. The ST inhibitor also suppressed the interaction of myeloma cells with E-selectin, MADCAM1, and VCAM1 and altered the post-translational modification of α4 integrin. In effect, sialyltransferase inhibition restricted myeloma cells from entering the protective bone marrow (BM) microenvironment wherein chemotherapeutic agents work less efficacious [
148].
A recent development to the 3F
ax-Neu5Ac inhibitor is the preparation of C-5-modified 3-fluoro sialic acid analogues [
149]. Taking into consideration that the potency element of the compound depends on N-substitution, they replaced the natural N-acetamide group with a carbamate functionality which resulted in novel inhibitors
60–
67 with much improved inhibitory activities and prolonged sialylation inhibition (
Table 7). Very interestingly, all carbamate analogues
62–
67 with facile modification were more potent than compound
60. Compound
61 and
62 only differ in the α-substituent but the carbamate analogue exhibited a 56-fold increase in potency. This improvement was attributed to the more efficient metabolism of the designed inhibitors to their active CMP analogues, which in turn increased the effective inhibitor concentration inside the cells. It is worth mentioning that in spite of such potencies, there was no significant preference observed for the inhibition of α2,3-linked over α2,6-linked sialylation [
149]. Nonetheless, further in vivo biological studies involving these new set of cell permeable ST inhibitors remain warranted. To this date, the actual downstream targets of these sugar-based inhibitors continue to be unresolved.
3.2. Non-Carbohydrate or Non-Sugar-Based ST Inhibitors
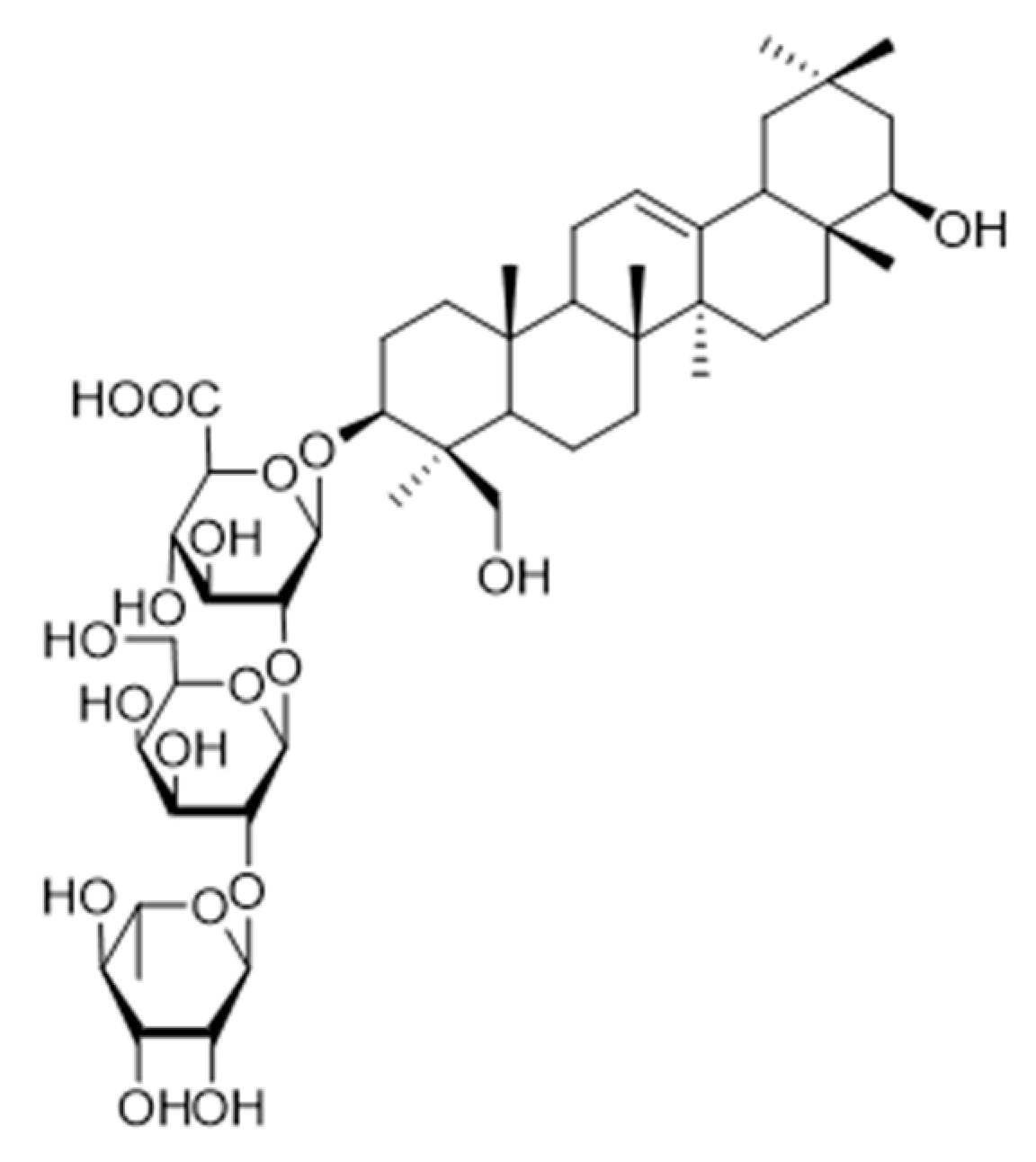
One of the current challenges in developing effective ST inhibitors for clinical applications remains to be the poor membrane permeability of most small molecule candidates previously discussed. ST inhibitors obtained from natural sources are usually more lipophilic but are highly limited because of the lengthy screening processes and tedious chiral separations that are required to identify and purify desired targets. Despite these difficulties, Tsai and coworkers reported that soyasaponin I (
Figure 12), a natural product derived from crude soybean saponin, which was identified by using a random screening process involving 7500 samples, acted as a highly specific inhibitor of ST3Gal-I (K
i = 2.3 μM) [
150]. In addition, soyasaponin I selectively depressed mRNA expression of ST3Gal-IV and reduced tumor cell surface α2,3-sialic acid expression, resulting in the modification of the invasive behavior of tumor cells. In a highly metastatic cancer cell line B16F10, soyasaponin I effectively and specifically attenuated α2,3-sialylation on the cell surface, inhibited the migration ability of cancer cells, and enhanced cell adhesion to extracellular matrix proteins [
151,
152]. Treatment with soyasaponin I was found to cause a reduction of lung metastasis in mice, suggesting that the natural product altered sialylation of cell surface adhesion molecules bringing about a significant reduction in the ability of tumor cells to distribute to the lungs.
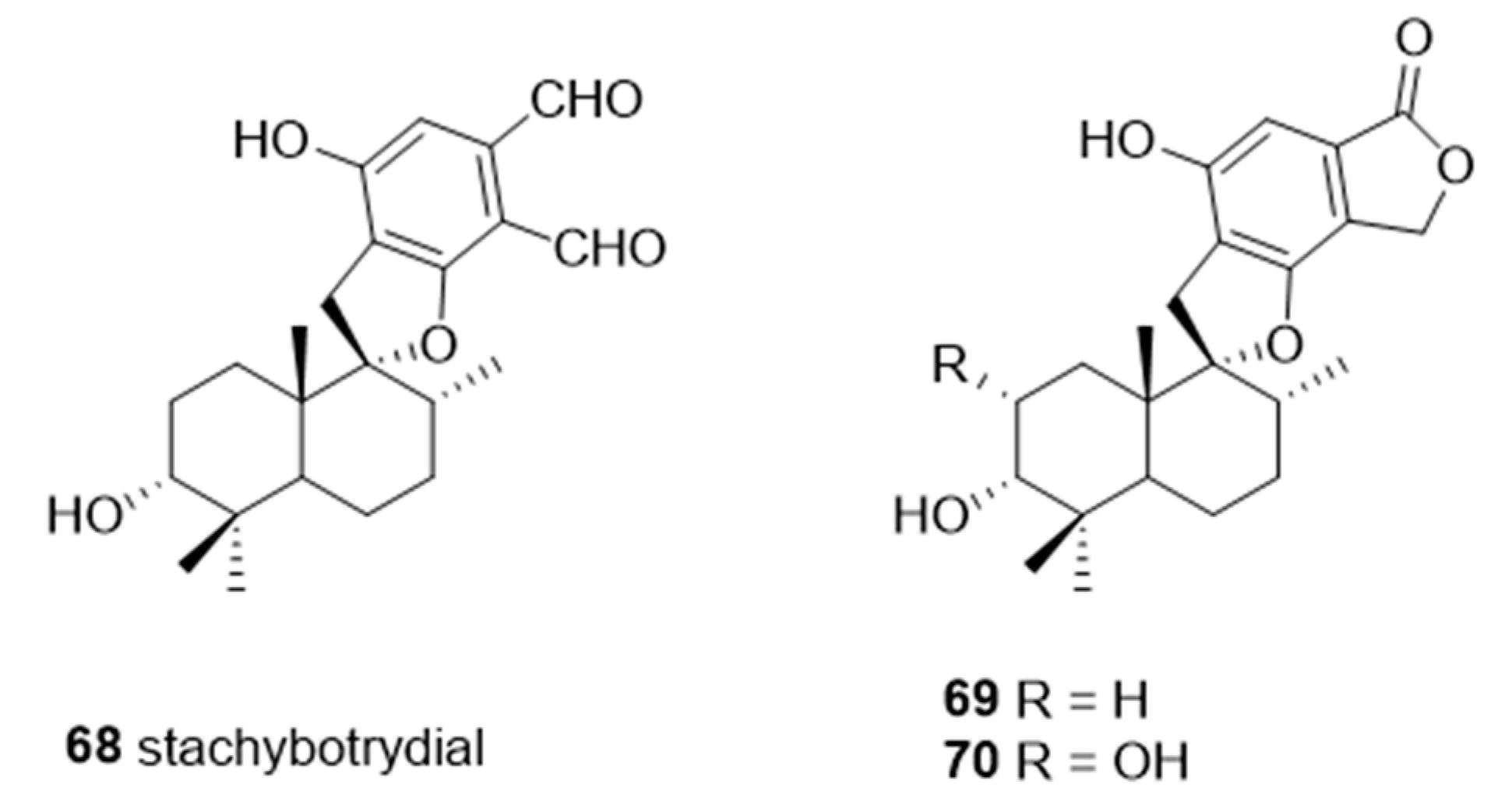
A naturally occurring member of the spirocyclic drimane family, stachybotrydial
68, was isolated from the fungal strain,
Stachybotrys cylindrospora, in 1993 [
153]. Studies showed that
68 has inhibitory activities against cholesterol esterase [
154], inositol monophosphatase [
155], and avian myeloblastosis virus protease [
156]. Among the spirocyclic drimanes tested,
68–70 (
Figure 13) were shown to play diverse roles in inhibition of TNFα liberation from macrophage and antiplasmodial or antiviral activities [
157,
158]. Interestingly, the results of a study of sialyltransferase inhibition showed that
68–
70 are potent inhibitors against various STs with IC
50 values in the micromolar range (
Table 8) [
159]. These findings suggest that spirocyclic drimanes, and especially stachybotrydial, represent an interesting scaffold for the development of human ST inhibitors but may be impeded by their broad range of off-target effects and considerable synthetic inaccessibility.
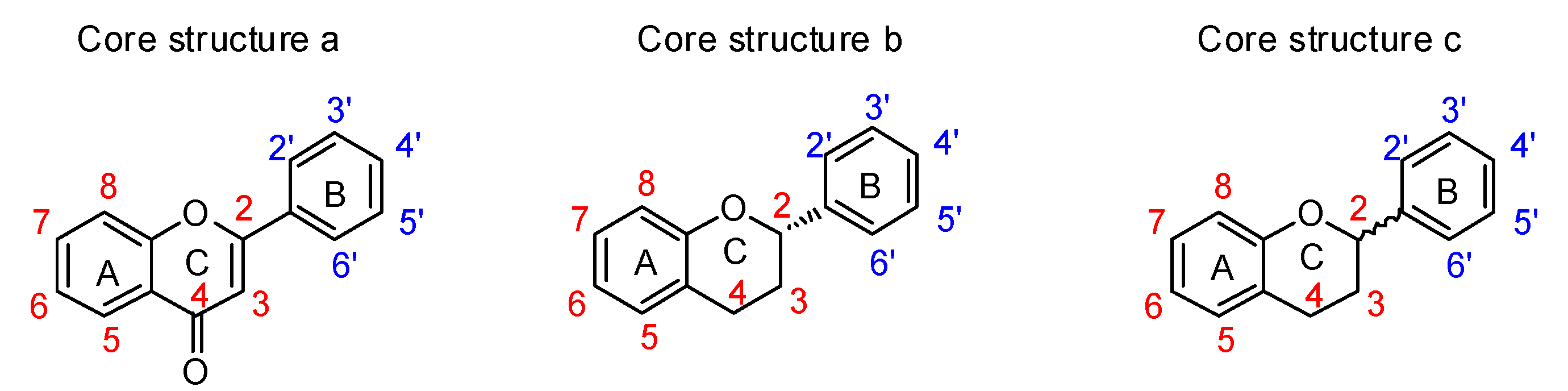
The utility of plant-derived flavonoids [
160] as anti-tumor [
161,
162], anti-inflammatory [
163], antibacterial [
164], and antioxidant agents [
165] has also been recognized. Members of this inhibitor family possess structures containing two phenyl groups (A-ring and B-ring) connected through three carbon atoms and one oxygen atom as part of a tetrahydropyran or dihydropyranone C-ring (
Figure 14). Their broad range of biological activities suggest that flavonoids might also influence cancer-related processes. Suzuki and coworkers reported several flavonoid derivatives, containing three types of core structure (core a–c in
Figure 14) and their results showed that some of these flavonoid analogues displayed significant inhibitory effects against rat ST6Gal-I, human ST6Gal-I, and rat ST3Gal-III (
Table 9) [
166]. Among the tested substances,
77 and
79 displayed the highest inhibitory activities, with IC
50 values of 1.4–7.3 μM against the three STs.
Interestingly, the data revealed that most flavonoid analogues that exhibited ST inhibitory activity commonly have the benzopyranone core a, rather than core b or c framework, indicating that a double bond between C2–C3 in the C-ring (
Figure 14) is essential for inhibitory activity. Furthermore, increasing the number of hydroxyl group on the B-ring of these substances, as exemplified by
77 versus
79 (
Table 9), increased inhibitory activity. This observation implies that increasing the hydrophilic character of the B-ring leads to an enhancement of binding to sialyltransferases. In contrast, flavonoid analogues that contain a glucose group at C4′ of the B-ring are not ST inhibitors, suggesting that hydrophilic substituents larger than a hydroxyl group on the B-ring of the flavonoid backbone have an adverse effect on binding to STs. In addition, incorporation of a hydrophobic group such as methyl on A-ring hydroxyl groups of the flavonoids slightly enhanced inhibitory activity.
In view of the inhibitory activities displayed by flavonoids against sialyltransferases, the same group carried out studies to elucidate the nature of the inhibition against human ST6Gal-I by selected members of this series, including
71,
76, and
77. A summary of the kinetic parameters arising from this effort is given in
Table 10. The K
m value of recombinant human ST6Gal-I with CMP-Neu5Ac as the substrate was similar to that of the native enzyme reported elsewhere [
167]. The K
m values of ST6Gal-I for CMP-Neu5Ac increased from 4.77 to 5.21 μM in the presence of 30 μM
71 and to 25.6 μM in the presence of 80 μM
71. Studies using 75 μM
76 and 6 μM
77 showed the same trends in binding affinity (
Table 10). In the presence of
71,
76, or
77, the maximum rate of reaction (V
max) values of ST6Gal-I decreased in a dose-dependent manner. These kinetic data suggest that these flavonoid analogues displayed a mixed type of inhibition of the sialyltransferases. Based on these findings, the flavonoid analogues discovered by Suzuki and coworkers are particularly attractive and may warrant further biological studies due to their lipophilic character, membrane permeability, and drug-like heteroaromatic scaffold.
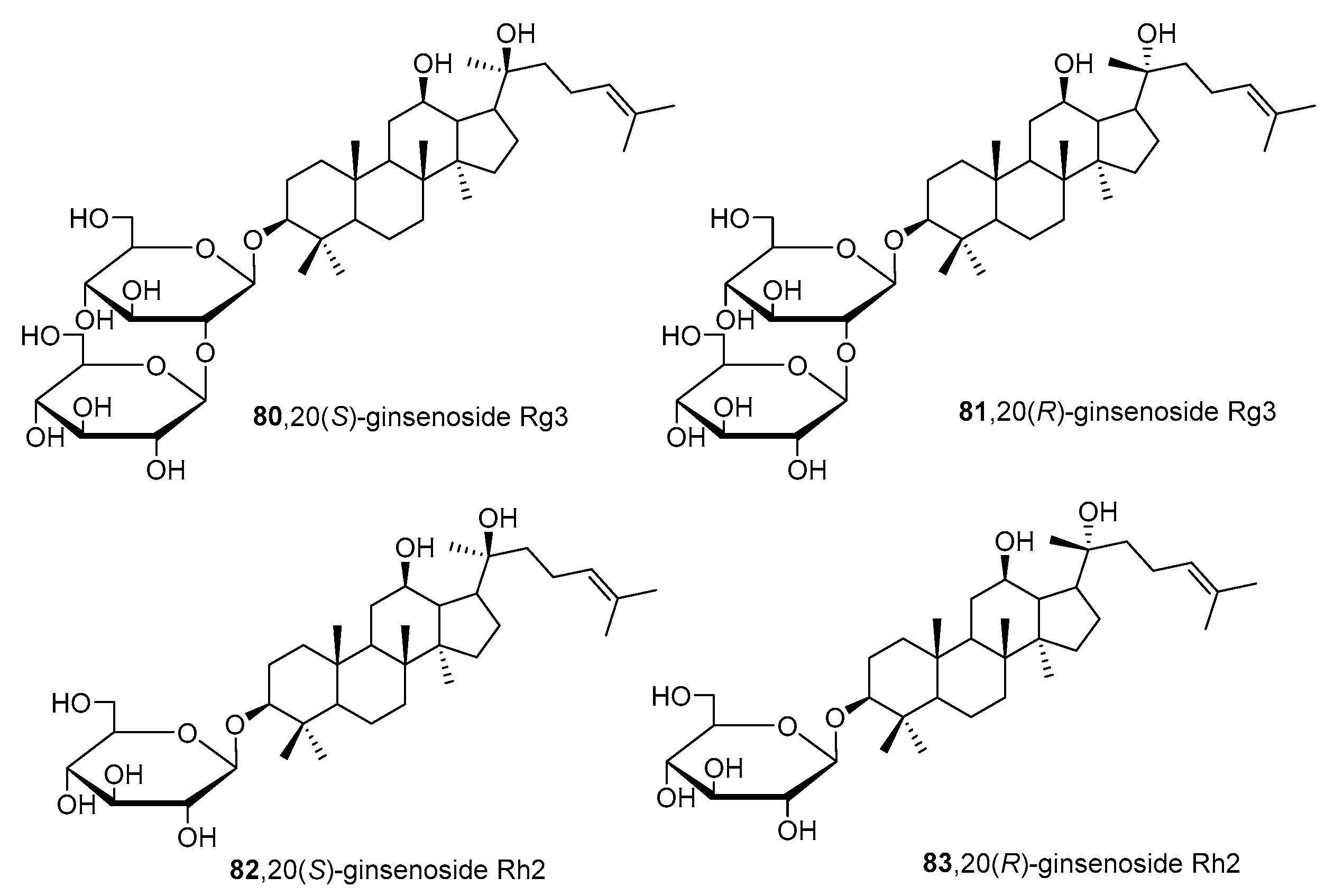
Recently, ginsenosides
80–
83 (
Figure 15) isolated from the ethanolic extract of
Panax ginseng C.A. Mey leaves were able to suppress total and free sialic acid expressions dose-dependently as well as inhibit ST expression [
168]. From flow cytometry analysis, all compounds blocked sialylation of α2,3- and α2,6-linked sialic acids in HepG2 cells. In detail, compounds
82 and
83 with only one monosaccharide group possessed stronger inhibition attributed to their higher lipophilicity and permeability. Moreover, the
R diastereomers were more inhibitory than their corresponding
S counterparts, which was attributed to the difference in planarity which could have affected how the substrate attaches to the active site. Data further revealed that these ginsenosides showed partial selective inhibition of α2,6-sialylation compared to α2,3-sialylation [
168]. However, further development of such natural product-based ST inhibitors remains limited by synthetic accessibility and availability.
Human bile acids (BAs), including primary BAs (cholic acid, chenodeoxycholic acid) and secondary BAs (deoxycholic acid, lithocholic acid), are oxidative metabolites of cholesterol in hepatocytes that form amphipathic derivatives with detergent-like, steroidal structures [
169]. The results of several studies clearly showed that BAs not only modulated their own biosynthesis and their secretion through intracellular bile acid signaling pathways but that they also played an important role in other metabolic pathways. Bile acid receptors have been recognized to be targets for drug development, as exemplified by the observation that BAs have pro-apoptotic and pro-inflammatory activities [
170]. Based on the fact that BAs showed in vivo potency in models for treatment of metabolic disorders, chronic liver diseases, hepatocellular cancer, and inflammatory diseases, we have examined the inhibition of STs involved in lung metastasis by lithocholic acid and other bile acids. This effort was inspired by the results of the studies with soyasaponin I, which possesses a pentacyclic ring system that is similar to the main skeleton of steroidal compounds. Epiandrosterone succinyl ester, a potent
Schistosoma japonicum glutathione
S-transferase inhibitor [
171], and lithocholic acid, a substrate of nuclear pregnane X receptor [
172], were identified through random screening of a library of steroidal compounds and shown to exhibit moderate inhibitory activities against ST3Gal-I with IC
50 values of 350 and 21 μM, respectively. Our investigation has expanded the group of substances displaying ST inhibitory activity to include the epiandrosterone derivatives
84–
86 and lithocholic acid derivatives
87–102. Inhibition constants of these substances towards ST3Gal-I are listed in
Table 11 [
173]. Among these bile acid analogues,
102 was observed to have the highest inhibitory activity towards ST3Gal-I, with an IC
50 of 5 μM. Analysis of a Lineweaver–Burk plot of the inhibition data showed that
102 is a noncompetitive inhibitor (K
i = 2.2 μM) towards the sialyl donor, cytidine monophosphate
N-acetylneuraminic acid (CMP-Neu5Ac). However, the mode of inhibition of
102 towards the sialylation acceptor has not been determined thus far. Overall, these observations provide insight into the ST inhibitory pattern of lithocholic acid derivatives arising from soyasaponin I. Furthermore, these studies have pushed forward the development of ST inhibitors by providing a new structural family of substances that have cell-permeable properties and potential selective inhibition capability.
We have further conducted in-depth biological studies on the Lith-O-Asp steroid
93 because it is more synthetically accessible than other lithocholic acid derivatives (
Table 11). Lith-O-Asp was found to exhibit inhibitory properties not only against ST3Gal-I, ST3Gal-III, and ST6Gal-I with IC
50 values in the low micromolar range (12–37 μM), but also against the migration and invasion abilities of various lung cancer cell lines [
174]. In addition, Lith-O-Asp also participated in anti-angiogenesis progression and decreased cell migration ability through inhibition of the integrin sialylation and downregulation of FAK/paxillin signaling pathway [
174].
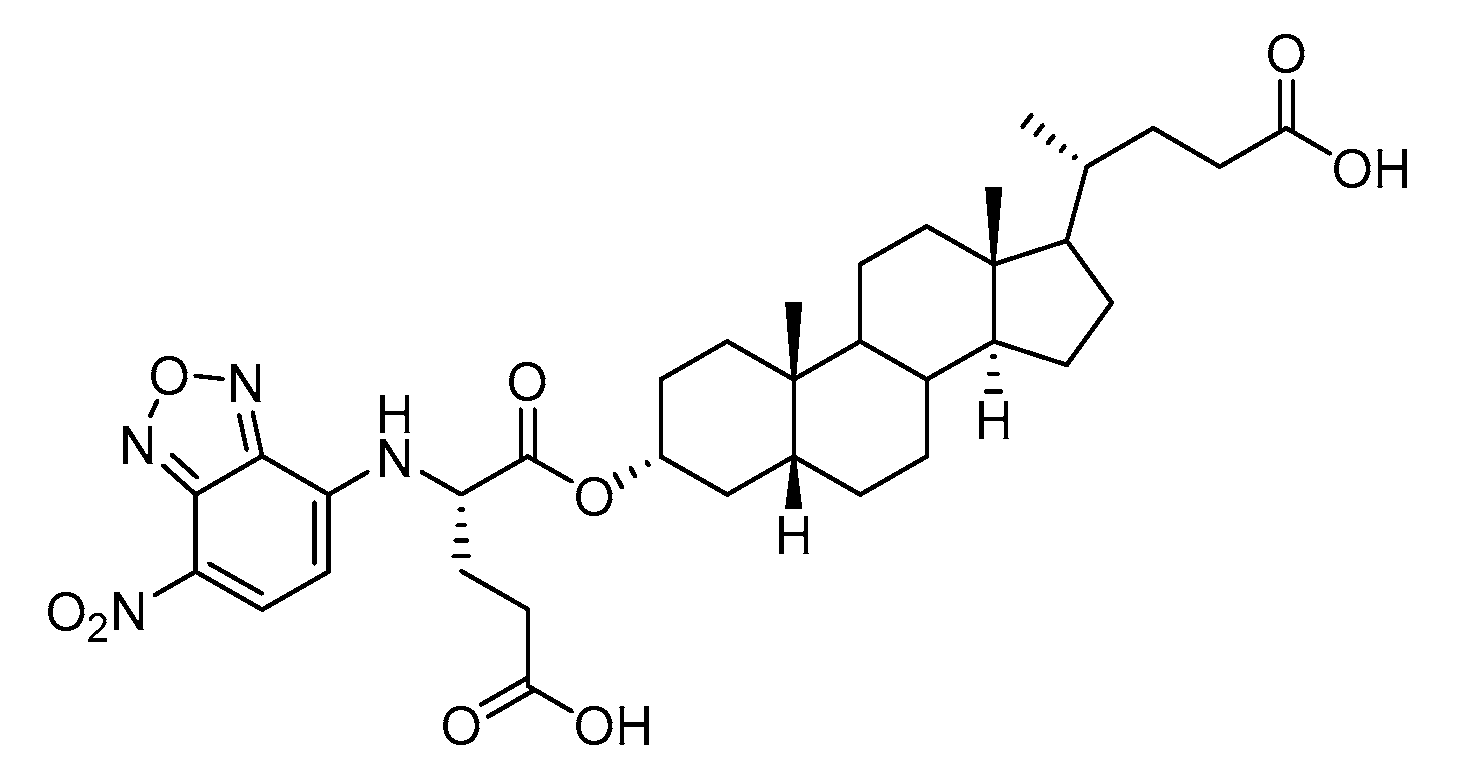
AL10 (
Figure 16), which possesses a lithocholic acid core structure containing a 7-nitro-2,1,3-benzoxadiazole (NBD) moiety, was initially discovered as a ST inhibitor in a structure-activity relationship (SAR) study. By effectively attenuating sialylation on the cell surface, cell-permeable AL10 inhibited adhesion, migration, actin polymerization, and invasion of α2,3-ST-overexpressing A549 and CL1-5 human lung cancer cells at nontoxic concentration levels [
175]. Furthermore, studies with this substance suggested that AL10-induced inhibition of adhesion and migration is associated with reduced sialylation of various integrins that led to attenuation of activation of the integrin downstream signaling mediator FAK. Importantly, AL10 effectively repressed lung metastasis in experimental animals without negatively affecting liver and kidney function, highlighting the pertinence of natural bile acids as an antimetastatic scaffold.
In our exploitation of the lithocholic acid skeleton, we have regioselectively synthesized B- and C-ring-modified analogues with improved ST inhibitory activities [
176]. Previous efforts have focused mainly on structural derivatization on the side chain carboxylic acid and the 3-α-OH groups, which led us to examine whether the steroid backbone influences the ST activity or not. In this particular work, we expanded the B and C rings via the insertion of -COO- or -CONH- to yield novel bile acid analogues
103–
108 with lactone or lactam functionalities (
Table 12). To our delight, compounds
103,
105, and
106 displayed greater potency with IC
50 values equal to or below 3 μM. Moreover, moderate selectivity for N-sialylation was observed for these set of analogues as α2,3-(O)-sialylation was not inhibited in concentrations as high as 100 μM. Given the potency and selectivity observed for these compounds, further structural optimization and modifications are currently underway.
In the previous subsection, tight-binding substrate analogues have been developed but are generally hampered for use in biological and clinical applications due to their low lipophilicity. Another major challenge in the development of ST inhibitors that must be addressed is subtype-selectivity. Hence, the discovery of cell-permeable inhibitors that can target specific ST isozymes in vivo remains crucial for the development of novel antimetastatic drugs with less off-target and side effects. In our most recent work, we took on this challenge and were able to synthesize and biologically evaluate a second-generation series of cell-permeable and N- versus O-selective ST inhibitors [
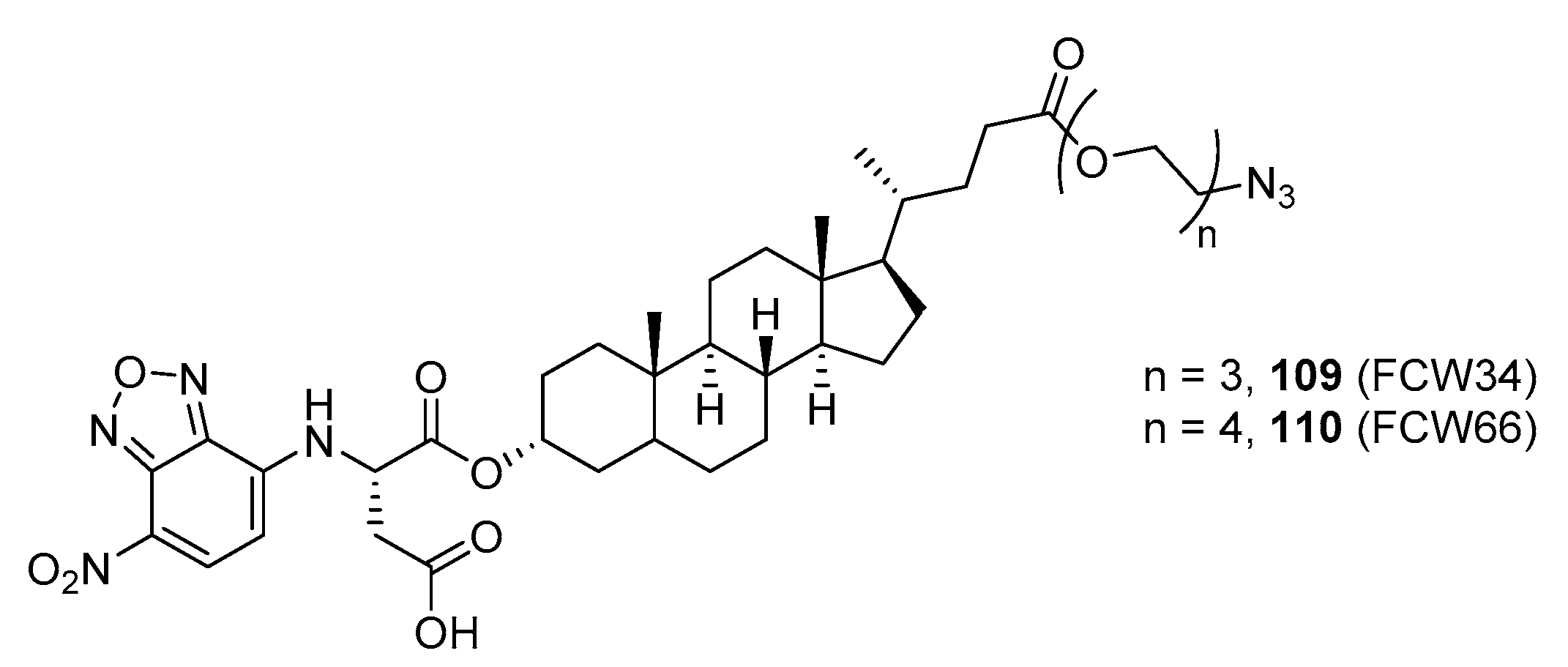
177]. Inspired by the previously reported pan-ST inhibitors (Lith-O-Asp and AL10) wherein we primarily focused on functionalization on the C3-α-OH group, we decided to divert our attention to derivatization at the cyclopentane ring side chain. To our delight, we discovered that conjugation of short-chain oligo(ethylene glycol) moieties to the carboxylic acid group led to a drastic improvement in subtype-selectivity (
Figure 17). Among this series, the two most promising compounds
109 (FCW34) and
110 (FCW66) exhibited submicromolar IC
50 values of 1.74 ± 0.09 μM and 1.01 ± 0.07 μM against α2,3-N-ST3Gal-III and 3.60 ± 0.40 μM and 4.90 ± 0.08 μM against α2,6-N-ST6Gal-I, respectively (
Table 13). In contrast, compounds
109 and
110 displayed no inhibition at 500 μM and only 50–60% inhibition at 1mM against α2,3-O-ST3Gal-I which is indicative of the isozyme selectivity towards the two
N-glycoprotein sialyltransferases. Further biological evaluation showed that the compounds inhibited breast cancer cell migration in a concentration-dependent manner, suppressed sialylation of integrin β subunits β1, β3, β4, and β5, hampered in vivo spontaneous metastasis, reduced in vivo tumor growth, and diminished angiogenic activity in vivo [
175]. To the extent of our knowledge, this is the first report of biologically extensively studied ST inhibitors with cell membrane permeability, ST isozyme selectivity, and antimetastatic properties, thereby representing a major milestone in the ST-targeted cancer drug discovery and development.
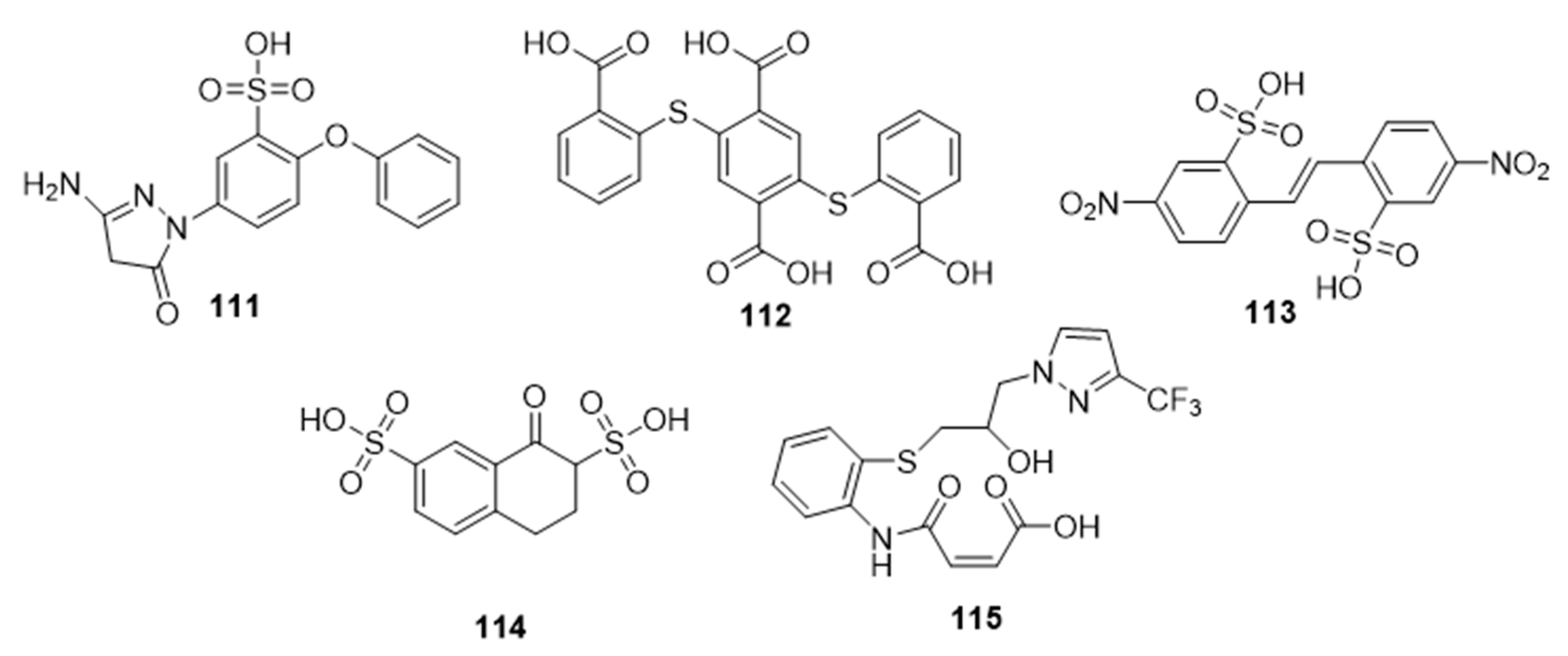
Facile and robust systems for high-throughput screening of sialyltransferase inhibitors that employ a fluorescence-polarization (FP)-based assay have also been developed by Paulson and coworkers [
178]. The library tested using this method contained the five thioether and/or sulfonic acid derivatives,
111–
115, which were found to be inhibitors of STs (
Figure 18,
Table 14). One member of this group,
111, exhibited potent inhibitory activity against ST3Gal-III with an IC
50 value of 3.1 μM and a similar inhibitory potency towards ST3Gal-I and ST6Gal-I (IC
50 = 14.1 and 10.8 μM, respectively) [
178]. Intriguingly,
115 exhibited selective inhibitory activity against ST3Gal-III with an IC
50 value of 1.7 μM, for it was a poor inhibitor of ST3Gal-I and ST6Gal-I (IC
50 > 500 μM). Their findings offered alternative, non-sugar, potentially selective, and drug-like scaffolds for new classes of ST inhibitors. However, validation of their therapeutic effects in vitro and in vivo remains lacking.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








































































