
Design, Synthesis, Biological Evaluation and In Silico Study of Benzyloxybenzaldehyde Derivatives as Selective ALDH1A3 Inhibitors

 , , , and
, , , and
Abstract
:1. Introduction
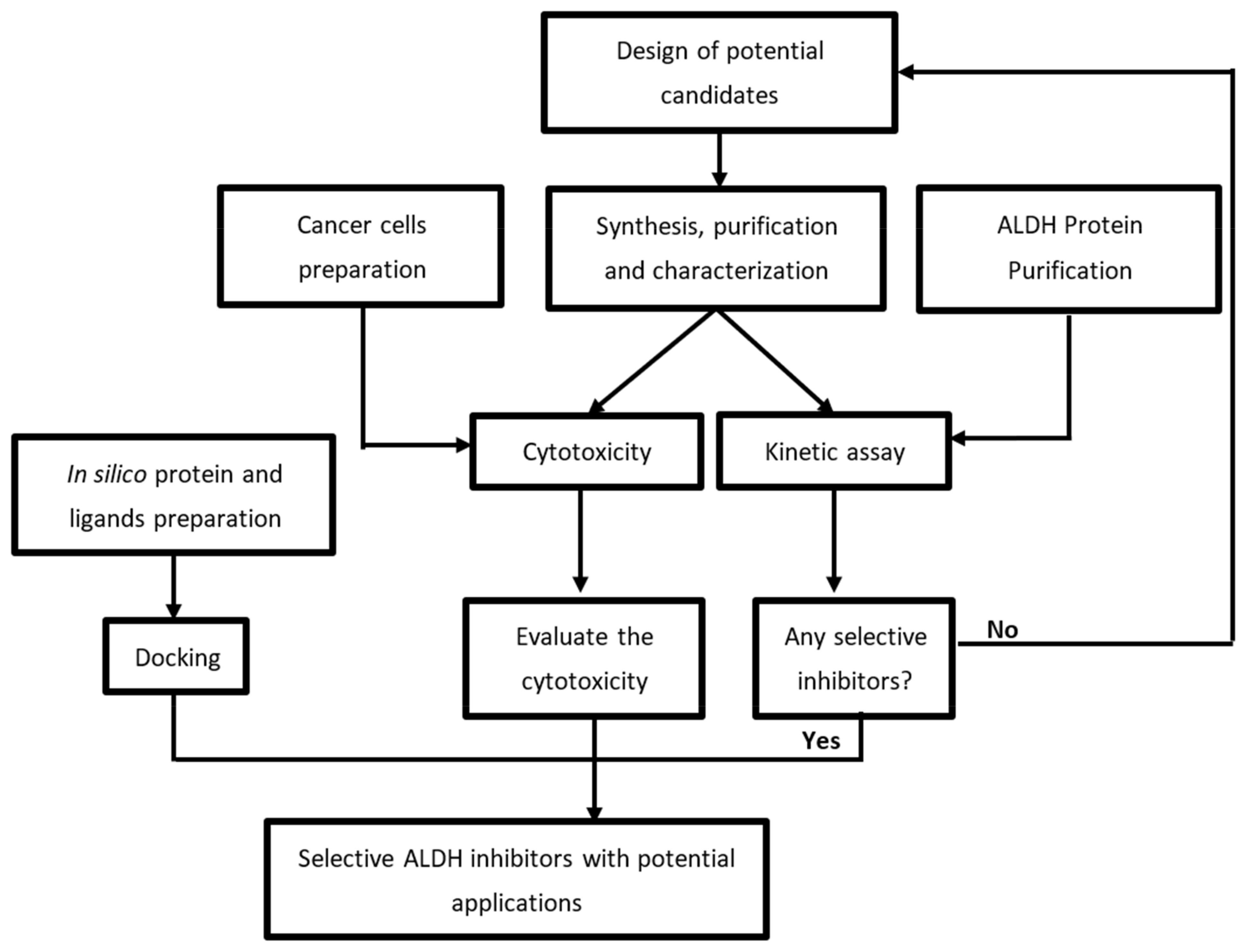
2. Results and Discussion
2.1. Chemistry
2.2. Inhibition Screening
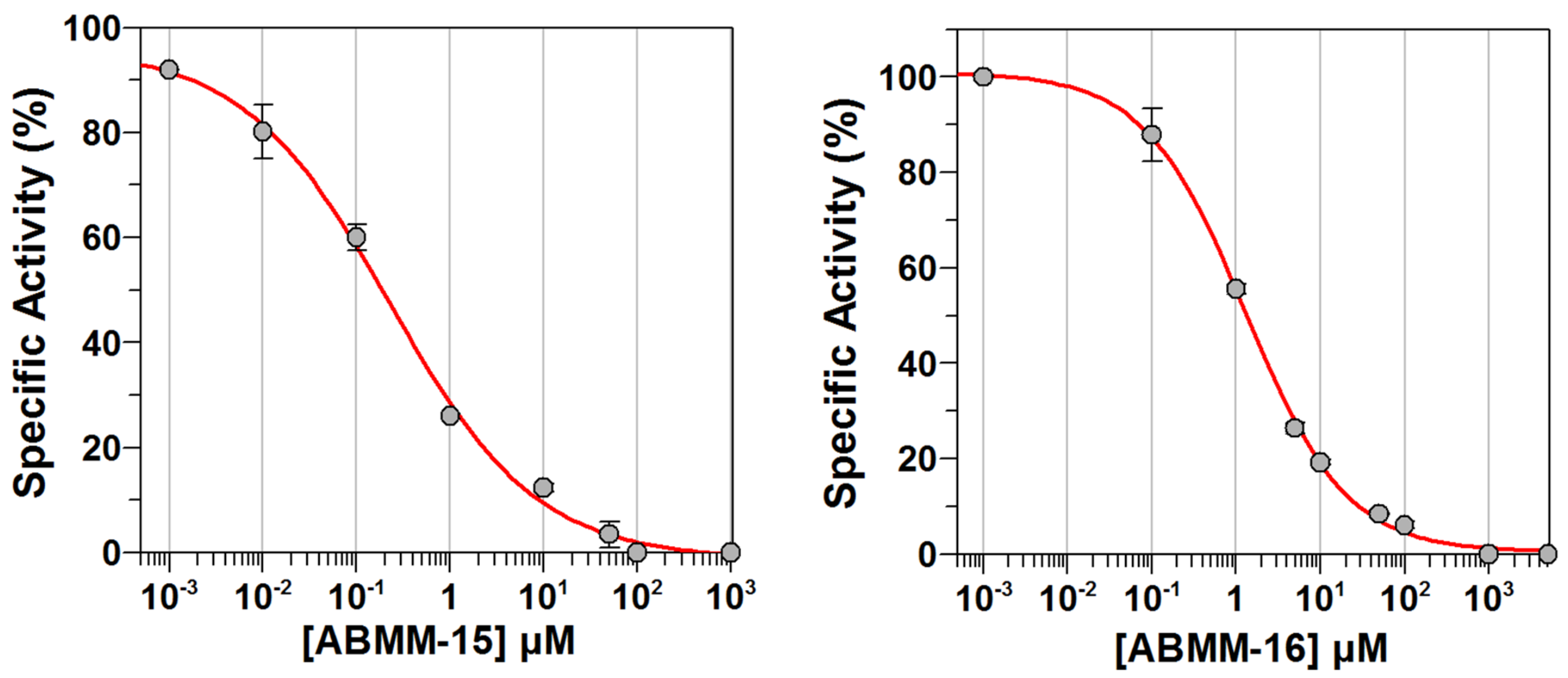
2.3. Determination of Half-Maximal Inhibitory Concentrations (IC50)
2.4. Cytotoxicity Experiments
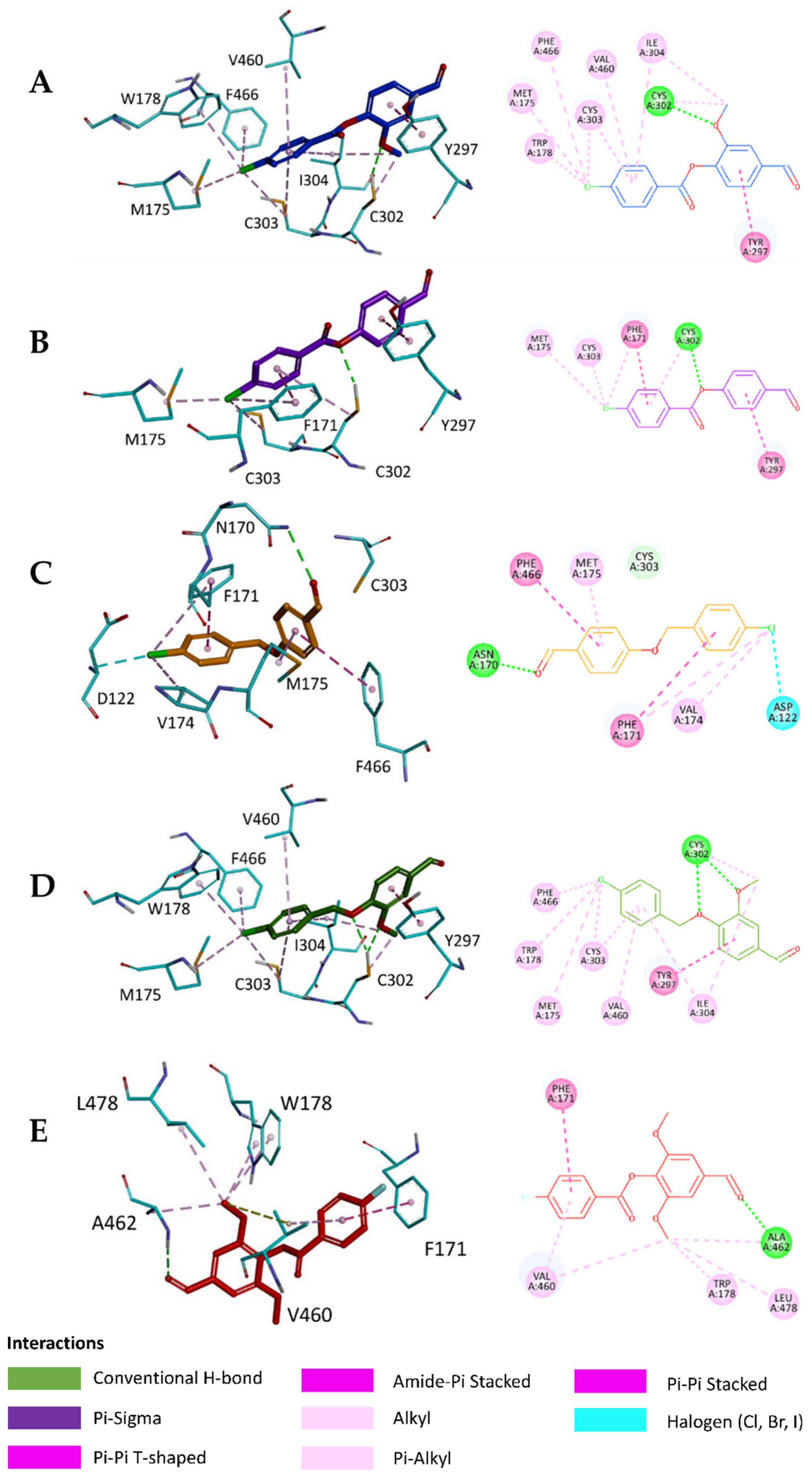
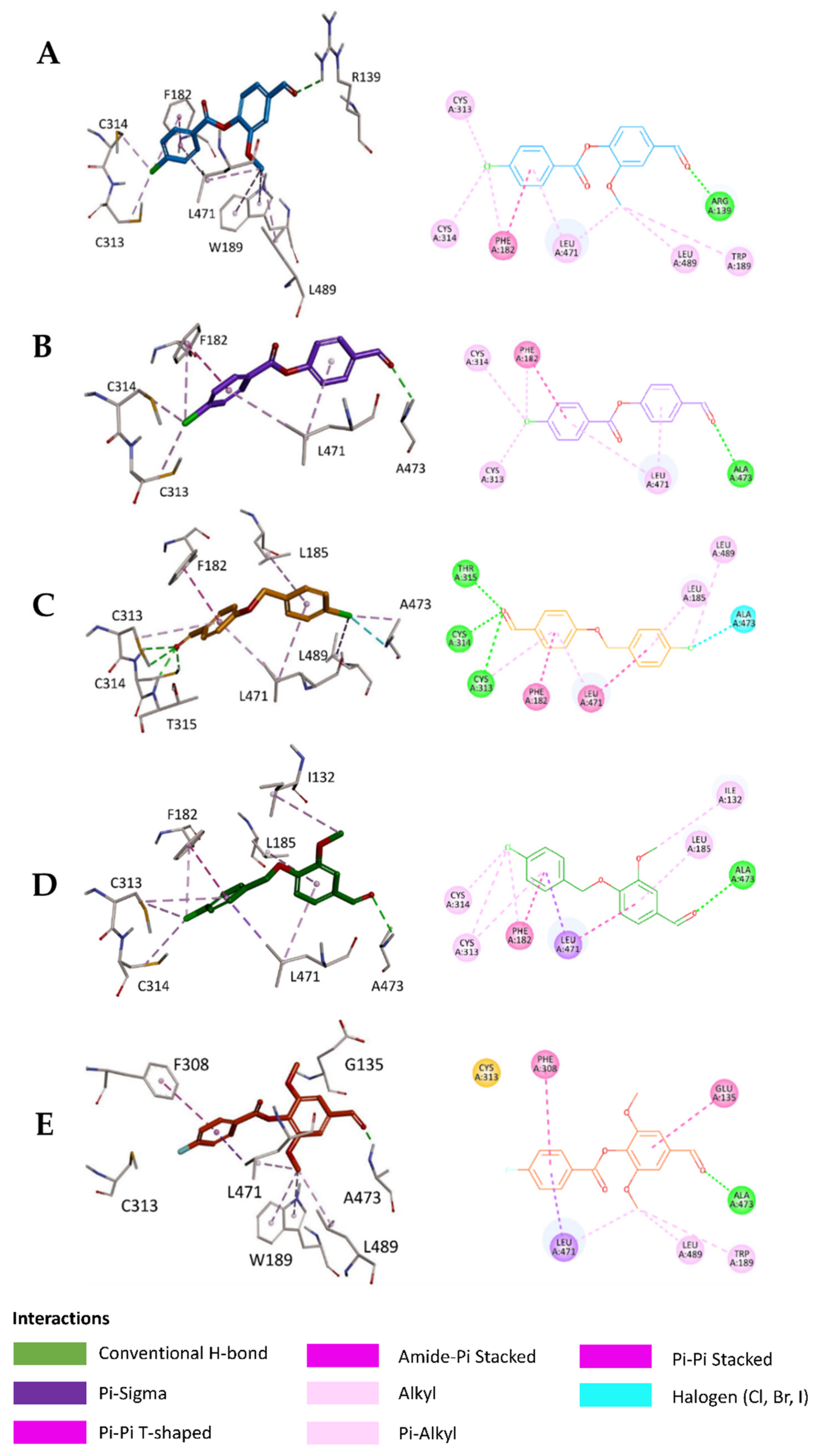
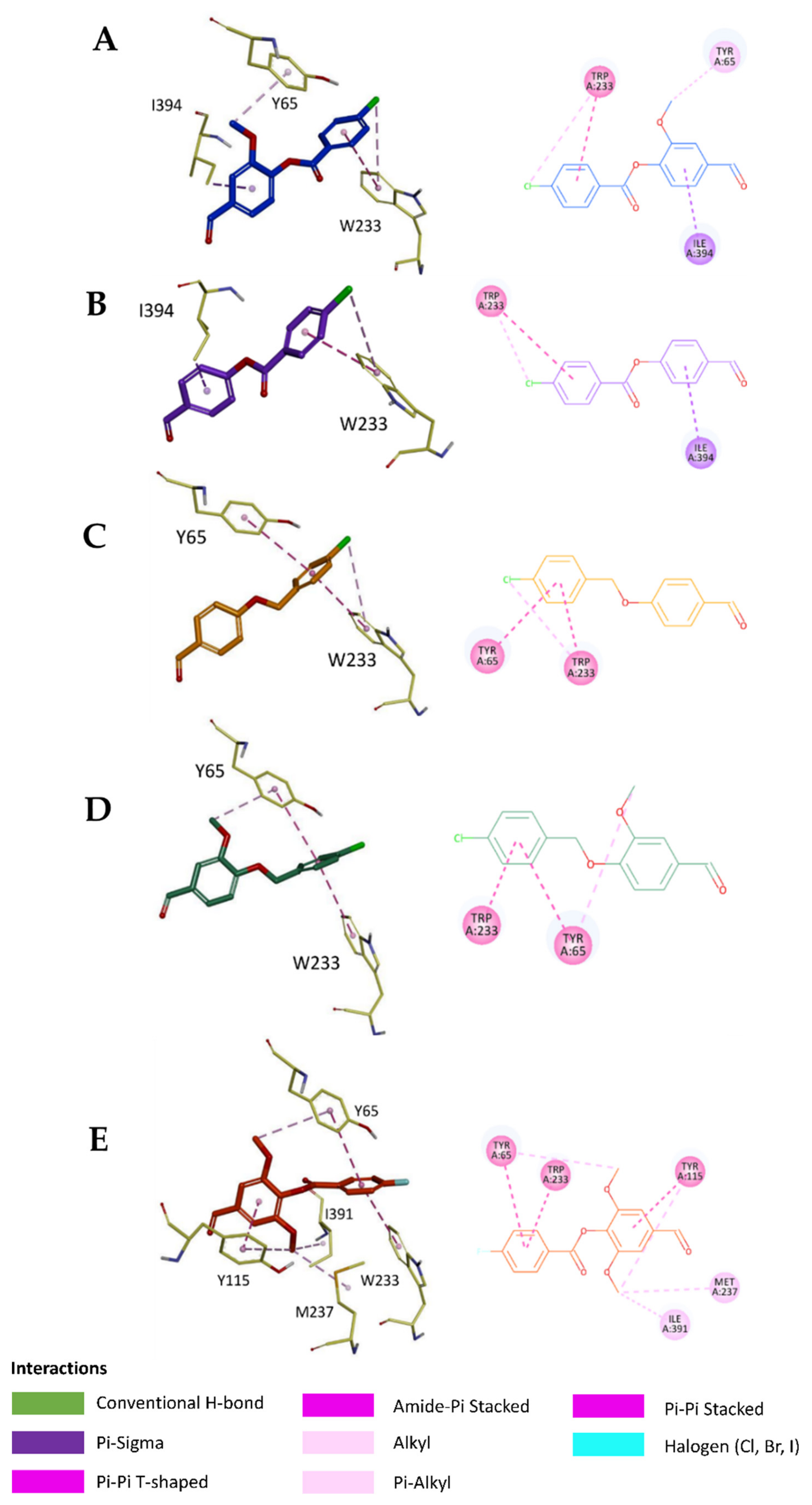
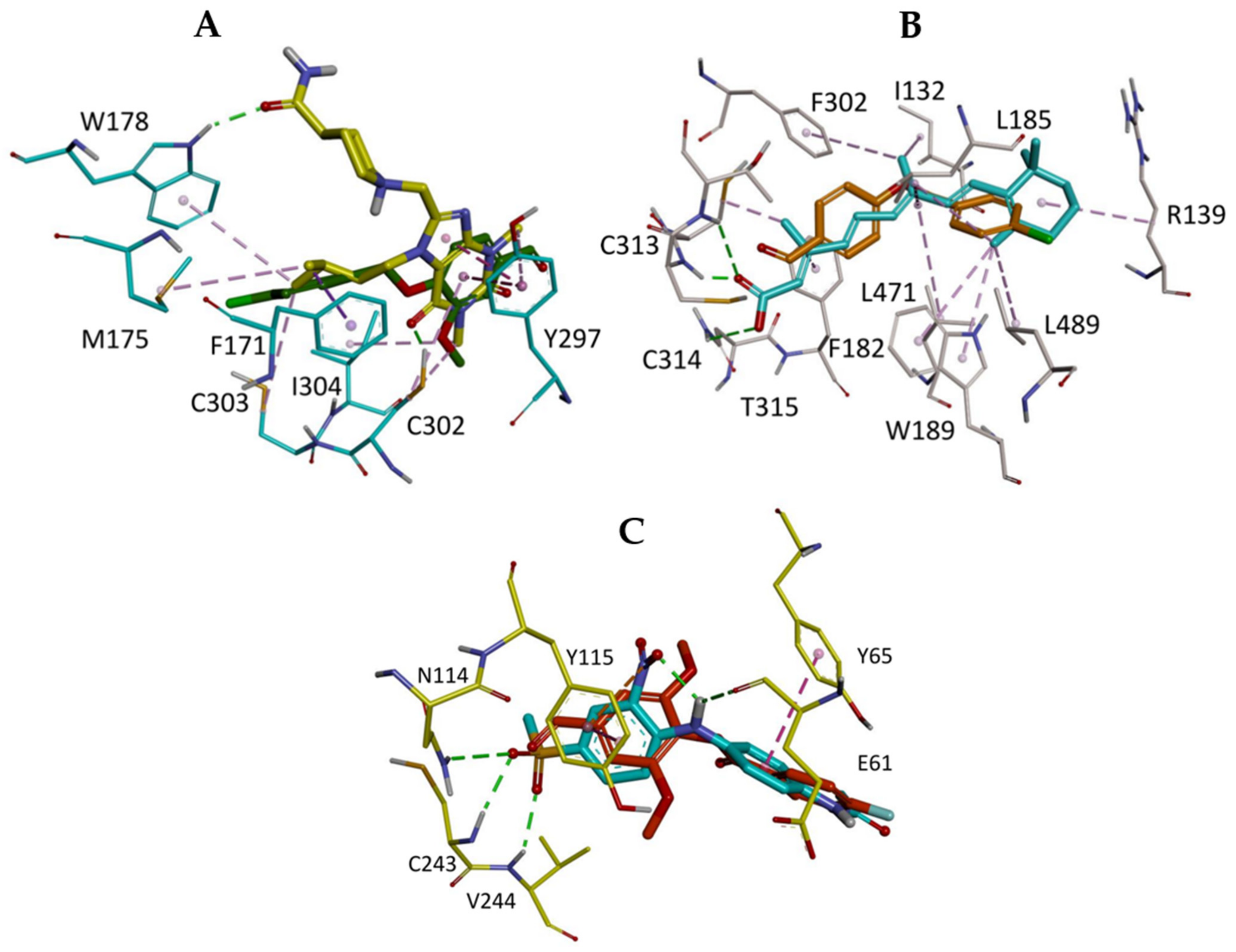
2.5. In Silico Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of Compounds ABMM-1–6, 17, 18, 27, 28, 32–34
3.1.2. General Procedure for the Synthesis of Compounds ABMM-15, 16, 24, 26
3.1.3. General Procedure for the Synthesis of Compounds ABMM-19, 23, 25, 35
4-Formyl-2-methoxyphenyl 4-Chlorobenzoate (ABMM-1)
4-Formylphenyl 4-Chlorobenzoate (ABMM-2)
4-Formylphenyl 4-Fluorobenzoate (ABMM-3)
4-Formylphenyl 4-Nitrobenzoate (ABMM-4)
4-Formyl-2-methoxyphenyl 4-Nitrobenzoate (ABMM-5)
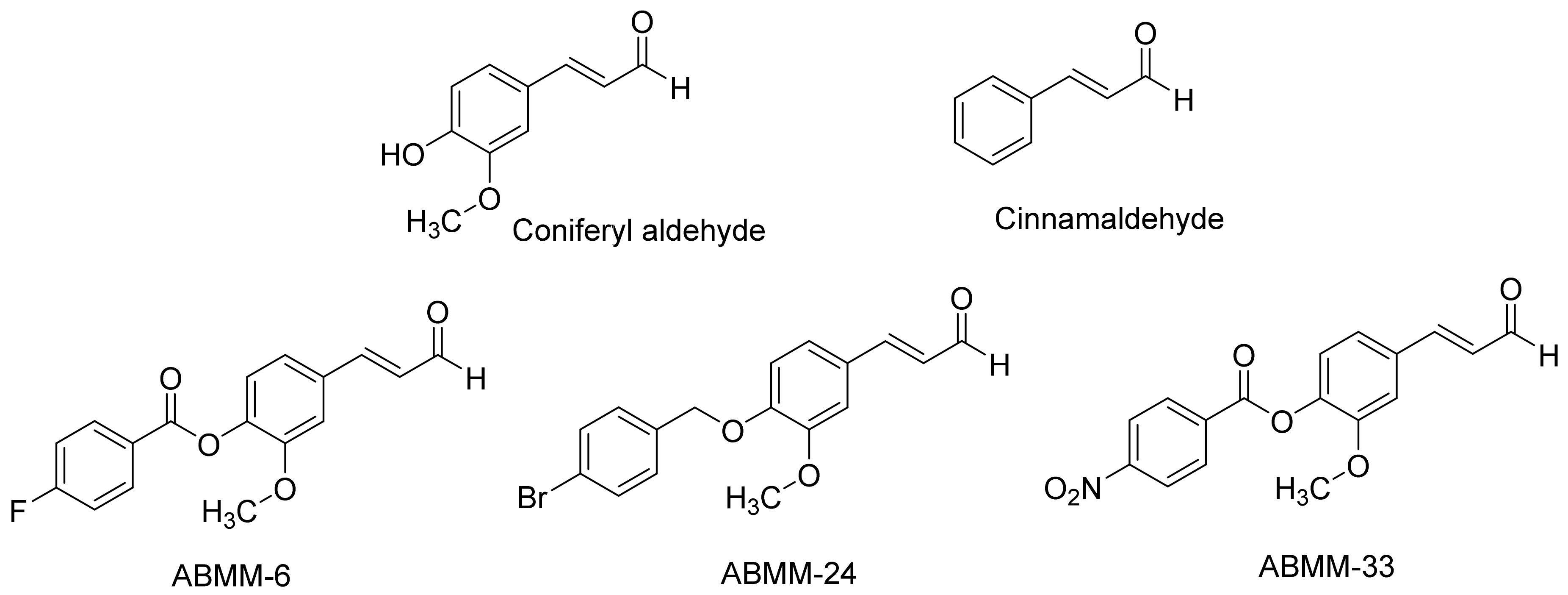
(E)-2-Methoxy-4-(3-oxoprop-1-en-1-yl)phenyl 4-Fluorobenzoate (ABMM-6)
4-((4-Chlorobenzyl)oxy)benzaldehyde (ABMM-15)
4-((4-Chlorobenzyl)oxy)-3-methoxybenzaldehyde (ABMM-16)
3-Formylphenyl 4-Fluorobenzoate (ABMM-17)
4-Formyl-2,6-dimethoxyphenyl 4-Fluorobenzoate (ABMM-18)
(E)-4-(3-Hydroxyprop-1-en-1-yl)-2-methoxyphenyl 4-Fluorobenzoate (ABMM-19)
4-(Hydroxymethyl)-2,6-dimethoxyphenyl 4-Fluorobenzoate (ABMM-23)
(E)-3-(4-((4-Bromobenzyl)oxy)-3-methoxyphenyl)acrylaldehyde (ABMM-24)
(E)-3-(4-((4-Bromobenzyl)oxy)-3-methoxyphenyl)prop-2-en-1-ol (ABMM-25)
(E)-3-(4-((4-Bromobenzyl)oxy)phenyl)acrylic Acid (ABMM-26)
(E)-3-(4-((4-Fluorobenzoyl)oxy)phenyl)acrylic Acid (ABMM-27)
4-Formyl-2-methoxyphenyl 4-(Chloromethyl)benzoate (ABMM-28)
4-Formylphenyl 4-(Chloromethyl)benzoate (ABMM-32)
(E)-2-Methoxy-4-(3-oxoprop-1-en-1-yl)phenyl 4-Nitrobenzoate (ABMM-33)
2-Formylphenyl 4-Chlorobenzoate (ABMM-34)
4-(Hydroxymethyl)phenyl 4-Chlorobenzoate (ABMM-35)
3.2. Purification of Recombinant Human ALDHs and Enzymatic Assays
3.3. Inhibition Screening
3.4. Determination of Half-Maximal Inhibitory Concentrations (IC50)
3.5. Biology
3.5.1. Cell Culture
3.5.2. Cytotoxicity Experiment
3.6. In Silico Study
3.6.1. Protein and Ligand Preparation
3.6.2. Molecular Docking
3.6.3. Visualization of the Docked Poses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Kelson, T.L.; McVoy, J.R.S.; Rizzo, W.B. Human liver fatty aldehyde dehydrogenase: Microsomal localization, purification, and biochemical characterization. Biochim. Biophys. Acta BBA Gen. Subj. 1997, 1335, 99–110. [Google Scholar] [CrossRef]
- Arnold, S.L.; Kent, T.; Hogarth, C.; Griswold, M.D.; Amory, J.; Isoherranen, N. Pharmacological inhibition of ALDH1A in mice decreases all-trans retinoic acid concentrations in a tissue specific manner. Biochem. Pharmacol. 2015, 95, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Ang, H.L.; Duester, G. Retinoic acid biosynthetic enzyme ALDH1 localizes in a subset of retinoid-dependent tissues during xenopus development. Dev. Dyn. 1999, 215, 264–272. [Google Scholar] [CrossRef]
- Albalat, R.; Cañestro, C. Identification of Aldh1a, Cyp26 and RAR orthologs in protostomes pushes back the retinoic acid genetic machinery in evolutionary time to the bilaterian ancestor. Chem. Interact. 2009, 178, 188–196. [Google Scholar] [CrossRef]
- Neuville, P.; Yan, Z.-q.; Gidlöf, A.; Pepper, M.S.; Hansson, G.K.; Gabbiani, G.; Sirsjö, A. Retinoic acid regulates arterial smooth muscle cell proliferation and phenotypic features in vivo and in vitro through an RARα-dependent signaling pathway. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, T.; Crandall, J.E.; Nanescu, S.E.; Quadro, L.; Shearer, K.; Ross, A.; McCaffery, P. Patterning of retinoic acid signaling and cell proliferation in the hippocampus. Hippocampus 2012, 22, 2171–2183. [Google Scholar] [CrossRef] [Green Version]
- Crowe, D.L.; Kim, R.; Chandraratna, R.A.S. Retinoic acid differentially regulates cancer cell proliferation via dose-dependent modulation of the mitogen-activated protein kinase pathway. Mol. Cancer Res. 2003, 1, 532–540. [Google Scholar] [PubMed]
- Karsy, M.; Albert, L.; Tobias, M.E.; Murali, R.; Jhanwar-Uniyal, M. All-trans retinoic acid modulates cancer stem cells of glioblastoma multiforme in an MAPK-dependent manner. Anticancer Res. 2010, 30, 4915–4920. [Google Scholar] [PubMed]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Torres, M.; Allan, A.L. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin. Exp. Metastasis 2016, 33, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Toledo-Guzmán, M.E.; Hernández, M.I.; Gómez-Gallegos, Á.A.; Ortiz-Sánchez, E. ALDH as a stem cell marker in solid tumors. Curr. Stem Cell Res. Ther. 2019, 14, 375–388. [Google Scholar] [CrossRef]
- Carpentino, J.E.; Hynes, M.J.; Appelman, H.D.; Zheng, T.; Steindler, D.A.; Scott, E.W.; Huang, E.H. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Res. 2009, 69, 8208–8215. [Google Scholar] [CrossRef] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.-M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2009, 16, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.-H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pors, K.; Moreb, J.S. Aldehyde dehydrogenases in cancer: An opportunity for biomarker and drug development? Drug Discov. Today 2014, 19, 1953–1963. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and Its expression is predictive of metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Servat, C.C.; Cardona, A.F.; Giménez-Capitán, A.; Drozdowskyj, A.; Berenguer, J.; Bracht, J.W.P.; Ito, M.; Karachaliou, N.; Rosell, R. Cancer stem cell biomarkers in EGFR-mutation-positive non-small-cell lung cancer. Clin. Lung Cancer 2019, 20, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Hu, H.; Huang, H.; Bao, Z.; Yang, P.; Wang, Y.; You, G.; Yan, W.; Jiang, T.; et al. ALDH1A3: A marker of mesenchymal phenotype in gliomas associated with cell invasion. PLoS ONE 2015, 10, e0142856. [Google Scholar] [CrossRef]
- Thomas, M.L.; de Antueno, R.; Coyle, K.; Sultan, M.; Cruickshank, B.M.; Giacomantonio, M.A.; Giacomantonio, C.A.; Duncan, R.; Marcato, P. Citral reduces breast tumor growth by inhibiting the cancer stem cell marker ALDH1A3. Mol. Oncol. 2016, 10, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Canino, C.; Luo, Y.; Marcato, P.; Blandino, G.; Pass, H.; Cioce, M. A STAT3-NFkB/DDIT3/CEBPβ axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget 2015, 6, 12637–12653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurth, I.; Hein, L.; Mäbert, K.; Peitzsch, C.; Koi, L.; Cojoc, M.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Cancer stem cell related markers of radioresistance in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 34494–34509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelardi, E.; Colombo, G.; Picarazzi, F.; Ferraris, D.; Mangione, A.; Petrarolo, G.; Aronica, E.; Rizzi, M.; Mori, M.; La Motta, C.; et al. A selective competitive inhibitor of aldehyde dehydrogenase 1A3 hinders cancer cell growth, invasiveness and stemness in vitro. Cancers 2021, 13, 356. [Google Scholar] [CrossRef]
- Pan, M.; Li, M.; You, C.; Zhao, F.; Guo, M.; Xu, H.; Li, L.; Wang, L.; Dou, J. Inhibition of breast cancer growth via miR-7 suppressing ALDH1A3 activity concomitant with decreasing breast cancer stem cell subpopulation. J. Cell. Physiol. 2020, 235, 1405–1416. [Google Scholar] [CrossRef] [Green Version]
- Khanna, M.; Chen, C.-H.; Kimble-Hill, A.; Parajuli, B.; Perez-Miller, S.; Baskaran, S.; Kim, J.; Dria, K.; Vasiliou, V.; Mochly-Rosen, D.; et al. Discovery of a novel class of covalent inhibitor for aldehyde dehydrogenases. J. Biol. Chem. 2011, 286, 43486–43494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskar, A.A.; Alam Khan, M.; Ahmad, S.; Hashmi, A.; Younus, H. Citral inhibition of human salivary aldehyde dehydrogenase. Cell Biophys. 2019, 78, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Gates, K.S.; Henzl, M.T.; Tanner, J.J. Diethylaminobenzaldehyde is a covalent, irreversible inactivator of ALDH7A1. ACS Chem. Biol. 2015, 10, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Farrés, J.; Wang, T.T.Y.; Cunningham, S.J.; Weiner, H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry 1995, 34, 2592–2598. [Google Scholar] [CrossRef]
- Kopečný, D.; Tylichová, M.; Snégaroff, J.; Popelková, H.; Šebela, M. Carboxylate and aromatic active-site residues are determinants of high-affinity binding of ω-aminoaldehydes to plant aminoaldehyde dehydrogenases. FEBS J. 2011, 278, 3130–3139. [Google Scholar] [CrossRef] [Green Version]
- Moretti, A.; Li, J.; Donini, S.; Sobol, R.W.; Rizzi, M.; Garavaglia, S. Crystal structure of human aldehyde dehydrogenase 1A3 complexed with NAD+ and retinoic acid. Sci. Rep. 2016, 6, 35710. [Google Scholar] [CrossRef] [Green Version]
- Pequerul, R.; Vera, J.; Giménez-Dejoz, J.; Crespo, I.; Coines, J.; Porté, S.; Rovira, C.; Parés, X.; Farrés, J. Structural and kinetic features of aldehyde dehydrogenase 1A (ALDH1A) subfamily members, cancer stem cell markers active in retinoic acid biosynthesis. Arch. Biochem. Biophys. 2020, 681, 108256. [Google Scholar] [CrossRef]
- Riveros-Rosas, H.; González-Segura, L.; Julián-Sánchez, A.; Díaz-Sanchez, Á.; Muñoz-Clares, R.A. Structural determinants of substrate specificity in aldehyde dehydrogenases. Chem. Interact. 2013, 202, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Bian, Y.; Zhou, X.; Yuan, Q.; Wei, S.; Xue, L.; Yang, F.; Dong, Q.; Wang, W.; Zheng, B.; et al. A small-molecule activator of mitochondrial aldehyde dehydrogenase 2 reduces the severity of cerulein-induced acute pancreatitis. Biochem. Biophys. Res. Commun. 2019, 522, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, J.-Y.; Hong, K.H.; Mikles, D.C.; Georg, G.I.; Goldstein, A.S.; Amory, J.; Schonbrunn, E. Structural basis of ALDH1A2 inhibition by irreversible and reversible small molecule inhibitors. ACS Chem. Biol. 2017, 13, 582–590. [Google Scholar] [CrossRef]
- Parajuli, B.; Georgiadis, T.M.; Fishel, M.L.; Hurley, T.D. Development of selective inhibitors for human aldehyde dehydrogenase 3A1 (ALDH3A1) for the enhancement of cyclophosphamide cytotoxicity. ChemBioChem 2014, 15, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Morgan, C.A.; Parajuli, B.; Buchman, C.D.; Dria, K.; Hurley, T.D. N,N-diethylaminobenzaldehyde (DEAB) as a substrate and mechanism-based inhibitor for human ALDH isoenzymes. Chem. Interact. 2015, 234, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, H.; Hu, J.H.; Sanny, C.G. Rate-limiting steps for the esterase and dehydrogenase reaction catalyzed by horse liver aldehyde dehydrogenase. J. Biol. Chem. 1976, 251, 3853–3855. [Google Scholar] [CrossRef]
- Rodríguez-Zavala, J.S.; Allali-Hassani, A.; Weiner, H. Characterization of E. coli tetrameric aldehyde dehydrogenases with atypical properties compared to other aldehyde dehydrogenases. Protein Sci. 2006, 15, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, B.; Kimble-Hill, A.C.; Khanna, M.; Ivanova, Y.; Meroueh, S.; Hurley, T.D. Discovery of novel regulators of aldehyde dehydrogenase isoenzymes. Chem. Interact. 2011, 191, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Jung, K.-H.; Lee, J.H.; Moon, S.H.; Cho, Y.S.; Lee, K.-H. Inhibition of aldehyde dehydrogenase 1 enhances the cytotoxic effect of retinaldehyde on A549 cancer cells. Oncotarget 2017, 8, 99382–99393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreb, J.S.; Zucali, J.R.; Ostmark, B.; Benson, N.A. Heterogeneity of aldehyde dehydrogenase expression in lung cancer cell lines is revealed by Aldefluor flow cytometry-based assay. Cytom. Part B Clin. Cytom. 2007, 72B, 281–289. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Liu, L.-Q.; He, Y.-L.; Kong, W.-J.; Huang, S.-A. Cytotoxic effect of trans-cinnamaldehyde on human leukemia K562 cells. Acta Pharmacol. Sin. 2010, 31, 861–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najar, B.; Shortrede, J.; Pistelli, L.; Buhagiar, J. Chemical composition and in vitro cytotoxic screening of sixteen commercial essential oils on five cancer cell lines. Chem. Biodivers. 2019, 17, e1900478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Liu, S.-L.; Qi, M.-H.; Zou, X. Cinnamaldehyde/chemotherapeutic agents interaction and drug-metabolizing genes in colorectal cancer. Mol. Med. Rep. 2013, 9, 669–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Baek, S.H. Combination therapy with cinnamaldehyde and hyperthermia induces apoptosis of A549 non-small cell lung carcinoma cells via regulation of reactive oxygen species and mitogen-activated protein kinase family. Int. J. Mol. Sci. 2020, 21, 6229. [Google Scholar] [CrossRef]
- Hosseinzadeh, L.; Abbasi, A.; Hajialyani, M.; Jalilian, F.; Yaghmaei, P.; Navid, S.J.; Motamed, H. Evaluation of the cytotoxic and apoptogenic effects of cinnamaldehyde on U87MG cells alone and in combination with doxorubicin. Res. Pharm. Sci. 2020, 15, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Cabello, C.M.; Bair, W.B.; Lamore, S.D.; Ley, S.; Bause, A.S.; Azimian, S.; Wondrak, G.T. The cinnamon-derived Michael acceptor cinnamic aldehyde impairs melanoma cell proliferation, invasiveness, and tumor growth. Free. Radic. Biol. Med. 2009, 46, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Sudanich, S.; Tiyaworanant, S.; Yenjai, C. Cytotoxicity of flavonoids and isoflavonoids from Crotalaria bracteata. Nat. Prod. Res. 2017, 31, 2641–2646. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res. Treat. 2011, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Sołobodowska, S.; Giebultowicz, J.; Wolinowska, R.; Wroczyński, P. Contribution of ALDH1A1 isozyme to detoxification of aldehydes present in food products. Acta Pol. Pharm. Drug Res. 2013, 69, 1380–1383. [Google Scholar]
- Markossian, S.; Grossman, A.; Brimacombe, K.; Arkin, M.; Auld, D.; Austin, C.P.; Baell, J.; Chung, T.D.Y.; Coussens, N.P.; Dahlin, J.L.; et al. Assay Guidance Manual; Eli Lilly & Company: Indianapolis, IN, USA; National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliand, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Morgan, C.A.; Hurley, T. Characterization of two distinct structural classes of selective aldehyde dehydrogenase 1A1 inhibitors. J. Med. Chem. 2015, 58, 1964–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | Remaining Activity ± SE (%) at 10 µM Compound | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ALDH1A1 | ALDH1A3 | ALDH3A1 | |||||||
| R1 | R2 | R3 | X | Y | [S] at Saturation (30 µM Hex) | [S] near Km (10 µM Hex) | [S] at Saturation (250 µM Hex) | [S] at Saturation (250 µM 4-NBA) | |
| ABMM-1 | Cl | OCH3 | H | C=O | CHO | 81.93 ± 2.9 | 21.07 ± 0.1 | 61.14 ± 2.6 | ND |
| ABMM-2 | Cl | H | H | C=O | CHO | 56.98 ± 5.8 | 94.76 ± 8.3 | 79.70 ± 1.9 | 162.08 ± 10.7 |
| ABMM-5 | NO2 | OCH3 | H | C=O | CHO | 92.38 ± 4.6 | 61.93 ± 5.8 | 79.78 ± 5.9 | ND |
| ABMM-6 | F | OCH3 | H | C=O | CH=CHCHO | 94.76 ± 8.3 | 76.26 ± 3.5 | 91.27 ± 1.8 | 92.54 ± 11.4 |
| ABMM-15 | Cl | H | H | CH2 | CHO | 48.22 ± 2.8 | 0.14 ± 0.1 | 12.35 ± 0.7 | 146.35 ± 2.7 |
| ABMM-16 | Cl | OCH3 | H | CH2 | CHO | 42.27 ± 1.7 | 4.27 ± 1.8 | 19.22 ± 0.5 | 117.43 ± 3.5 |
| ABMM-17 * | Cl | H | --- | C=O | CHO | 57.85 ± 5.5 | 113.0 ± 0.5 | 95.06 ± 1.9 | 104.43 ± 2.4 |
| ABMM-18 | F | OCH3 | OCH3 | C=O | CHO | 70.86 ± 2.6 | 16.01 ± 3.4 | 77.30 ± 9.9 | 96.69 ± 3.3 |
| ABMM-19 | F | OCH3 | H | C=O | CH=CHCH2OH | 89.13 ± 0.4 | 88.32 ± 0.7 | 77.91 ± 7.4 | 124.32 ± 9.3 |
| ALDH1A3 Remaining Activity ± SE (%) at 10 µM Compound | ||
|---|---|---|
| Pre-Incubation | 5 min | 20 min |
| ABMM-15 | 12.35 ± 0.72 | 13.10 ± 4.35 |
| ABMM-16 | 19.22 ± 0.51 | 18.88 ± 1.81 |

| Compound | (O) Position Relative To (Y) | R1 | R2 | R3 | X | Y | Toxicity (IC50 (µM) ± SE) | IC50 (µM) ± SE ALDH1A3 | |
|---|---|---|---|---|---|---|---|---|---|
| A549 | H1299 | ||||||||
| ABMM-1 | para | Cl | OCH3 | H | C=O | CHO | >60 | >60 | ND |
| ABMM-2 | para | Cl | H | H | C=O | CHO | >60 | >60 | ND |
| ABMM-3 | para | F | H | H | C=O | CHO | >60 | >60 | ND |
| ABMM-4 | para | NO2 | H | H | C=O | CHO | ND | ND | ND |
| ABMM-5 | para | NO2 | OCH3 | H | C=O | CHO | ND | ND | ND |
| ABMM-6 | para | F | OCH3 | H | C=O | CH=CHCHO | >60 | 14 ± 2 | ND |
| ABMM-15 | para | Cl | H | H | CH2 | CHO | >60 | >60 | 0.23 ± 0.05 |
| ABMM-16 | para | Cl | OCH3 | H | CH2 | CHO | >60 | >60 | 1.29 ± 0.10 |
| ABMM-17 | meta | Cl | H | --- | C=O | CHO | >60 | 56 ± 2.65 | ND |
| ABMM-18 | para | F | OCH3 | OCH3 | C=O | CHO | >60 | >60 | ND |
| ABMM-19 | para | F | OCH3 | H | C=O | CH=CHCH2OH | >60 | 46.33 ± 4.48 | ND |
| ABMM-23 | para | F | OCH3 | OCH3 | C=O | CH2OH | 51 ± 8 | >60 | ND |
| ABMM-24 | para | Br | OCH3 | H | CH2 | CH=CHCHO | >60 | 13.67 ± 1.67 | ND |
| ABMM-25 | para | Br | OCH3 | H | CH2 | CH=CHCH2OH | >60 | >60 | ND |
| ABMM-26 | para | Br | OCH3 | H | CH2 | CH=CHCOOH | >60 | >60 | ND |
| ABMM-27 | para | F | OCH3 | H | C=O | CH=CHCOOH | >60 | >60 | ND |
| ABMM-28 | para | CH2Cl | OCH3 | H | C=O | CHO | 50 ± 1 | 37.5 ± 1.5 | ND |
| ABMM-32 | para | CH2Cl | H | H | C=O | CHO | 51 ± 5 | 13 ± 2.08 | ND |
| ABMM-33 | para | NO2 | OCH3 | H | C=O | CH=CHCHO | >60 | 35.33 ± 1.76 | ND |
| ABMM-34 | ortho | Cl | H | H | C=O | CHO | >60 | >60 | ND |
| ABMM-35 | para | Cl | H | H | C=O | CH2OH | >60 | >60 | ND |
| Docking Scores (kcal/mol) | |||
|---|---|---|---|
| ALDH1A1 | ALDH1A3 | ALDH3A1 | |
| ABMM-1 | −7.709 | −7.717 | −7.238 |
| ABMM-2 | −7.104 | −7.844 | −5.826 |
| ABMM-15 | −7.246 | −7.804 | −6.707 |
| ABMM-16 | −8.157 | −8.306 | −7.029 |
| ABMM-18 | −6.193 | −7.753 | −7.402 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, A.I.M.; Ikhmais, B.; Batlle, E.; AbuHarb, W.K.; Jha, V.; Jaradat, K.T.; Jiménez, R.; Pequerul, R.; Parés, X.; Farrés, J.; et al. Design, Synthesis, Biological Evaluation and In Silico Study of Benzyloxybenzaldehyde Derivatives as Selective ALDH1A3 Inhibitors. Molecules 2021, 26, 5770. https://doi.org/10.3390/molecules26195770
Ibrahim AIM, Ikhmais B, Batlle E, AbuHarb WK, Jha V, Jaradat KT, Jiménez R, Pequerul R, Parés X, Farrés J, et al. Design, Synthesis, Biological Evaluation and In Silico Study of Benzyloxybenzaldehyde Derivatives as Selective ALDH1A3 Inhibitors. Molecules. 2021; 26(19):5770. https://doi.org/10.3390/molecules26195770
Chicago/Turabian StyleIbrahim, Ali I. M., Balqis Ikhmais, Elisabet Batlle, Waed K. AbuHarb, Vibhu Jha, Khaled T. Jaradat, Rafael Jiménez, Raquel Pequerul, Xavier Parés, Jaume Farrés, and et al. 2021. "Design, Synthesis, Biological Evaluation and In Silico Study of Benzyloxybenzaldehyde Derivatives as Selective ALDH1A3 Inhibitors" Molecules 26, no. 19: 5770. https://doi.org/10.3390/molecules26195770
APA StyleIbrahim, A. I. M., Ikhmais, B., Batlle, E., AbuHarb, W. K., Jha, V., Jaradat, K. T., Jiménez, R., Pequerul, R., Parés, X., Farrés, J., & Pors, K. (2021). Design, Synthesis, Biological Evaluation and In Silico Study of Benzyloxybenzaldehyde Derivatives as Selective ALDH1A3 Inhibitors. Molecules, 26(19), 5770. https://doi.org/10.3390/molecules26195770