
Understanding the Adsorption of Peptides and Proteins onto PEGylated Gold Nanoparticles

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. PEGylation of Gold Nanoparticles and Protein Selection
2.2. Adsorption of Small Peptides onto PEGylated AuNPs
2.3. Adsorption of Larger Proteins onto PEGylated AuNPs
3. Discussion
4. Materials and Methods
4.1. Synthesis of Citrate-Stabilized Gold Nanoparticles
4.2. Protein Preparation
4.3. PEGylated Gold Nanoparticle Preparation
4.4. Transmission Electron Miscroscopy (TEM) Measurement of PEGylated AuNPs
4.5. NMR Adsorption Measurements
4.6. Dynamic Light Scattering
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Cano, C.; Carril, M. Recent Developments in the Design of Non-Biofouling Coatings for Nanoparticles and Surfaces. Int. J. Mol. Sci. 2020, 21, E1007. [Google Scholar] [CrossRef] [Green Version]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, L.; Alvarez-Puebla, R.A.; Pazos-Perez, N. Surface Modifications of Nanoparticles for Stability in Biological Fluids. Materials 2018, 11, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, H.A.; Magsam, A.W.; Tarudji, A.W.; Romanova, S.; Weber, L.; Gee, C.C.; Madsen, G.L.; Bronich, T.K.; Kievit, F.M. Evaluating differential nanoparticle accumulation and retention kinetics in a mouse model of traumatic brain injury via Ktrans mapping with MRI. Sci. Rep. 2019, 9, 16099. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-D.; Wu, D.; Shen, X.; Liu, P.-X.; Yang, N.; Zhao, B.; Zhang, H.; Sun, Y.-M.; Zhang, L.-A.; Fan, F.-Y. Size-dependent in vivo toxicity of PEG-coated gold nanoparticles. Int. J. Nanomed. 2011, 6, 2071–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazile, D.; Prud’homme, C.; Bassoullet, M.-T.; Marlard, M.; Spenlehauer, G.; Veillard, M. Stealth Me.PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system. J. Pharm. Sci. 1995, 84, 493–498. [Google Scholar] [CrossRef]
- Cho, W.-S.; Cho, M.; Jeong, J.; Choi, M.; Cho, H.-Y.; Han, B.S.; Kim, S.H.; Kim, H.O.; Lim, Y.T.; Chung, B.H.; et al. Acute toxicity and pharmacokinetics of 13 nm-sized PEG-coated gold nanoparticles. Toxicol. Appl. Pharmacol. 2009, 236, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, F.; Di, H.; Liu, X.; Zhang, P.; Zhou, W.; Liu, D. Cross-Linked Poly(ethylene glycol) Shells for Nanoparticles: Enhanced Stealth Effect and Colloidal Stability. Langmuir 2019, 35, 8799–8805. [Google Scholar] [CrossRef] [PubMed]
- Qie, Y.; Yuan, H.; von Roemeling, C.A.; Chen, Y.; Liu, X.; Shih, K.D.; Knight, J.A.; Tun, H.W.; Wharen, R.E.; Jiang, W.; et al. Surface modification of nanoparticles enables selective evasion of phagocytic clearance by distinct macrophage phenotypes. Sci. Rep. 2016, 6, 26269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, D.A.; Vanhecke, D.; Michen, B.; Blank, F.; Gehr, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J. Nanotechnol. 2014, 5, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernández, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface Functionalization of Nanoparticles with Polyethylene Glycol: Effects on Protein Adsorption and Cellular Uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.-S.; Cho, M.; Jeong, J.; Choi, M.; Han, B.S.; Shin, H.-S.; Hong, J.; Chung, B.H.; Jeong, J.; Cho, M.-H. Size-dependent tissue kinetics of PEG-coated gold nanoparticles. Toxicol. Appl. Pharmacol. 2010, 245, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, S.; Simon, J.; Paßlick, D.; Frey, M.-L.; Wagner, M.; Mailänder, V.; Crespy, D.; Landfester, K. Brush Conformation of Polyethylene Glycol Determines the Stealth Effect of Nanocarriers in the Low Protein Adsorption Regime. Nano Lett. 2021, 21, 1591–1598. [Google Scholar] [CrossRef]
- García, I.; Sánchez-Iglesias, A.; Henriksen-Lacey, M.; Grzelczak, M.; Penadés, S.; Liz-Marzán, L.M. Glycans as Biofunctional Ligands for Gold Nanorods: Stability and Targeting in Protein-Rich Media. J. Am. Chem. Soc. 2015, 137, 3686–3692. [Google Scholar] [CrossRef]
- Dai, Q.; Walkey, C.; Chan, W.C.W. Polyethylene Glycol Backfilling Mitigates the Negative Impact of the Protein Corona on Nanoparticle Cell Targeting. Angew. Chem. Int. Ed. 2014, 53, 5093–5096. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Niidome, T.; Yamagata, M.; Okamoto, Y.; Akiyama, Y.; Takahashi, H.; Kawano, T.; Katayama, Y.; Niidome, Y. PEG-modified gold nanorods with a stealth character for in vivo applications. J. Controlled Release 2006, 114, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.L.; Reuter, K.G.; Kai, M.P.; Herlihy, K.P.; Jones, S.W.; Luft, J.C.; Napier, M.; Bear, J.E.; DeSimone, J.M. PEGylated PRINT Nanoparticles: The Impact of PEG Density on Protein Binding, Macrophage Association, Biodistribution, and Pharmacokinetics. Nano Lett. 2012, 12, 5304–5310. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Del. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Li Volsi, A.; Jimenez de Aberasturi, D.; Henriksen-Lacey, M.; Giammona, G.; Licciardi, M.; Liz-Marzán, L.M. Inulin coated plasmonic gold nanoparticles as a tumor-selective tool for cancer therapy. J. Mater. Chem. B 2016, 4, 1150–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyewumi, M.O.; Yokel, R.A.; Jay, M.; Coakley, T.; Mumper, R.J. Comparison of cell uptake, biodistribution and tumor retention of folate-coated and PEG-coated gadolinium nanoparticles in tumor-bearing mice. J. Control. Release 2004, 95, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Spadavecchia, J.; Movia, D.; Moore, C.; Maguire, C.M.; Moustaoui, H.; Casale, S.; Volkov, Y.; Prina-Mello, A. Targeted polyethylene glycol gold nanoparticles for the treatment of pancreatic cancer: From synthesis to proof-of-concept in vitro studies. Int. J. Nanomed. 2016, 11, 791–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, E.; Araban, V.; Pelaz, B.; Alvarez, A.; Taboada, P.; Mahmoudi, M.; del Pino, P. Photothermal effects on protein adsorption dynamics of PEGylated gold nanorods. Appl. Mater. Today 2019, 15, 599–604. [Google Scholar] [CrossRef]
- Zhang, M.; Li, X.H.; Gong, Y.D.; Zhao, N.M.; Zhang, X.F. Properties and biocompatibility of chitosan films modified by blending with PEG. Biomaterials 2002, 23, 2641–2648. [Google Scholar] [CrossRef]
- Zalipsky, S. Chemistry of polyethylene glycol conjugates with biologically active molecules. Adv. Drug Del. Rev. 1995, 16, 157–182. [Google Scholar] [CrossRef]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B. Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Du, Y.; Jin, J.; Liang, H.; Jiang, W. Structural and Physicochemical Properties and Biocompatibility of Linear and Looped Polymer-Capped Gold Nanoparticles. Langmuir 2019, 35, 8316–8324. [Google Scholar] [CrossRef]
- Hristov, D.R.; Lopez, H.; Ortin, Y.; O’Sullivan, K.; Dawson, K.A.; Brougham, D.F. Impact of dynamic sub-populations within grafted chains on the protein binding and colloidal stability of PEGylated nanoparticles. Nanoscale 2021, 13, 5344–5355. [Google Scholar] [CrossRef]
- Partikel, K.; Korte, R.; Stein, N.C.; Mulac, D.; Herrmann, F.C.; Humpf, H.-U.; Langer, K. Effect of nanoparticle size and PEGylation on the protein corona of PLGA nanoparticles. Eur. J. Pharm. Biopharm. 2019, 141, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Blume, J.E.; Manning, W.C.; Troiano, G.; Hornburg, D.; Figa, M.; Hesterberg, L.; Platt, T.L.; Zhao, X.; Cuaresma, R.A.; Everley, P.A.; et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020, 11, 3662. [Google Scholar] [CrossRef]
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C.W. Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fan, Z.; Li, P.Y.; Deng, J.; Arhontoulis, D.C.; Li, C.Y.; Bowne, W.B.; Cheng, H. Dense and Dynamic Polyethylene Glycol Shells Cloak Nanoparticles from Uptake by Liver Endothelial Cells for Long Blood Circulation. ACS Nano 2018, 12, 10130–10141. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-D.; Huang, L. Pharmacokinetics and Biodistribution of Nanoparticles. Mol. Pharm. 2008, 5, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Vangala, K.; Vo, T.; Zhang, D.; Fitzkee, N.C. A Three-Step Model for Protein–Gold Nanoparticle Adsorption. J. Phys. Chem. C 2014, 118, 8134–8142. [Google Scholar] [CrossRef]
- Xu, J.X.; Alom, M.S.; Fitzkee, N.C. Quantitative Measurement of Multiprotein Nanoparticle Interactions Using NMR Spectroscopy. Anal. Chem. 2021, 93, 11982–11990. [Google Scholar] [CrossRef]
- Xu, J.X.; Fitzkee, N.C. Solution NMR of Nanoparticles in Serum: Protein Competition Influences Binding Thermodynamics and Kinetics. Front. Physiol. 2021, 12, 715419. [Google Scholar] [CrossRef]
- Siriwardana, K.; Gadogbe, M.; Ansar, S.M.; Vasquez, E.S.; Collier, W.E.; Zou, S.; Walters, K.B.; Zhang, D. Ligand Adsorption and Exchange on Pegylated Gold Nanoparticles. J. Phys. Chem. C 2014, 118, 11111–11119. [Google Scholar] [CrossRef]
- Tsai, D.-H.; Lu, Y.-F.; DelRio, F.W.; Cho, T.J.; Guha, S.; Zachariah, M.R.; Zhang, F.; Allen, A.; Hackley, V.A. Orthogonal analysis of functional gold nanoparticles for biomedical applications. Anal. Bioanal. Chem. 2015, 407, 8411–8422. [Google Scholar] [CrossRef]
- Xue, Y.; Li, X.; Li, H.; Zhang, W. Quantifying thiol–gold interactions towards the efficient strength control. Nat. Commun. 2014, 5, 4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inkpen, M.S.; Liu, Z.F.; Li, H.; Campos, L.M.; Neaton, J.B.; Venkataraman, L. Non-chemisorbed gold–sulfur binding prevails in self-assembled monolayers. Nat. Chem. 2019, 11, 351–358. [Google Scholar] [CrossRef]
- Rahme, K.; Chen, L.; Hobbs, R.G.; Morris, M.A.; O’Driscoll, C.; Holmes, J.D. PEGylated gold nanoparticles: Polymer quantification as a function of PEG lengths and nanoparticle dimensions. RSC Adv. 2013, 3, 6085–6094. [Google Scholar] [CrossRef] [Green Version]
- Leff, D.V.; Brandt, L.; Heath, J.R. Synthesis and Characterization of Hydrophobic, Organically-Soluble Gold Nanocrystals Functionalized with Primary Amines. Langmuir 1996, 12, 4723–4730. [Google Scholar] [CrossRef]
- Selvakannan, P.R.; Mandal, S.; Phadtare, S.; Pasricha, R.; Sastry, M. Capping of Gold Nanoparticles by the Amino Acid Lysine Renders Them Water-Dispersible. Langmuir 2003, 19, 3545–3549. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Wet chemical synthesis of silver nanorods and nanowires of controllable aspect ratio. Chem. Commun. 2001, 617–618. [Google Scholar] [CrossRef]
- Provorse, M.R.; Aikens, C.M. Binding of carboxylates to gold nanoparticles: A theoretical study of the adsorption of formate on Au20. Comput. Theor. Chem. 2012, 987, 16–21. [Google Scholar] [CrossRef]
- Krishna, M.M.G.; Hoang, L.; Lin, Y.; Englander, S.W. Hydrogen exchange methods to study protein folding. Methods 2004, 34, 51–64. [Google Scholar] [CrossRef]
- Perera, Y.R.; Hill, R.A.; Fitzkee, N.C. Protein Interactions with Nanoparticle Surfaces: Highlighting Solution NMR Techniques. Isr. J. Chem. 2019, 59, 962–979. [Google Scholar] [CrossRef] [PubMed]
- Szleifer, I. Protein Adsorption on Surfaces with Grafted Polymers: A Theoretical Approach. Biophys. J. 1997, 72, 595–612. [Google Scholar] [CrossRef] [Green Version]
- Unsworth, L.D.; Sheardown, H.; Brash, J.L. Protein-Resistant Poly(ethylene oxide)-Grafted Surfaces: Chain Density-Dependent Multiple Mechanisms of Action. Langmuir 2008, 24, 1924–1929. [Google Scholar] [CrossRef]
- Stanicki, D.; Larbanoix, L.; Boutry, S.; Vangijzegem, T.; Ternad, I.; Garifo, S.; Muller, R.N.; Laurent, S. Impact of the chain length on the biodistribution profiles of PEGylated iron oxide nanoparticles: A multimodal imaging study. J. Mater. Chem. B 2021, 9, 5055–5068. [Google Scholar] [CrossRef] [PubMed]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Frens, G. Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Link, S.; El-Sayed, M.A. Size and Temperature Dependence of the Plasmon Absorption of Colloidal Gold Nanoparticles. J. Phys. Chem. B 1999, 103, 4212–4217. [Google Scholar] [CrossRef]
- Jain, P.K.; Lee, K.S.; El-Sayed, I.H.; El-Sayed, M.A. Calculated Absorption and Scattering Properties of Gold Nanoparticles of Different Size, Shape, and Composition: Applications in Biological Imaging and Biomedicine. J. Phys. Chem. B 2006, 110, 7238–7248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Atwater, M.; Wang, J.; Huo, Q. Extinction coefficient of gold nanoparticles with different sizes and different capping ligands. Colloids Surf. B. Biointerfaces 2007, 58, 3–7. [Google Scholar] [CrossRef]
- Woods, K.E.; Perera, Y.R.; Davidson, M.B.; Wilks, C.A.; Yadav, D.K.; Fitzkee, N.C. Understanding Protein Structure Deformation on the Surface of Gold Nanoparticles of Varying Size. J. Phys. Chem. C 2016, 120, 27944–27953. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Perera, Y.R.; Davidson, M.B.; Fitzkee, N.C. Electrostatic Interactions and Protein Competition Reveal a Dynamic Surface in Gold Nanoparticle–Protein Adsorption. J. Phys. Chem. C 2016, 120, 24231–24239. [Google Scholar] [CrossRef] [Green Version]
- Jinasena, D.; Simmons, R.; Gyamfi, H.; Fitzkee, N.C. Molecular Mechanism of the Pin1–Histone H1 Interaction. Biochemistry 2019, 58, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
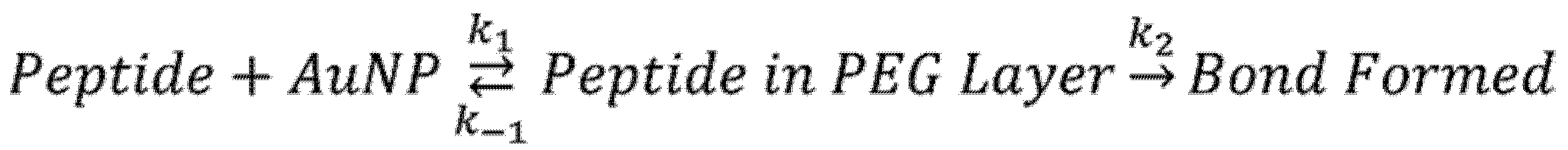{kind=link}
| AuNP-PEG Conjugate | DH (nm) | Zeta Potential (mV) | LSPR Peak (nm) | PEG Density (PEG/nm2) |
|---|---|---|---|---|
| AuNP | 18.4 ± 1.1 | −38 ± 3 | 520 | 0 |
| AuNP-PEG-5K | 27.7 ± 0.5 | −16.4 ± 0.8 | 523 | 0.96 ± 0.01 |
| AuNP-PEG-10K | 39 ± 2 | −22.1 ± 0.6 | 522 | 0.67 ± 0.01 |
| AuNP-PEG-30K | 78 ± 3 | −18 + 2 | 521 | 0.57 ± 0.01 |
| GSH | H1.5 | H1.5-Cys | WT GB3 | K19C GB3 | BSA | |
|---|---|---|---|---|---|---|
| Size (kDa) | 0.3 | 1.4 | 1.4 | 6.2 | 6.2 | 66 |
| Surface cysteines | 1 | 0 | 1 | 0 | 1 | 1 |
| Estimated net charge at pH 7 | −1 | 5 | 5 | −2 | −3 | −17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, Y.R.; Xu, J.X.; Amarasekara, D.L.; Hughes, A.C.; Abbood, I.; Fitzkee, N.C. Understanding the Adsorption of Peptides and Proteins onto PEGylated Gold Nanoparticles. Molecules 2021, 26, 5788. https://doi.org/10.3390/molecules26195788
Perera YR, Xu JX, Amarasekara DL, Hughes AC, Abbood I, Fitzkee NC. Understanding the Adsorption of Peptides and Proteins onto PEGylated Gold Nanoparticles. Molecules. 2021; 26(19):5788. https://doi.org/10.3390/molecules26195788
Chicago/Turabian StylePerera, Yasiru Randika, Joanna Xiuzhu Xu, Dhanush L. Amarasekara, Alex C. Hughes, Ibraheem Abbood, and Nicholas C. Fitzkee. 2021. "Understanding the Adsorption of Peptides and Proteins onto PEGylated Gold Nanoparticles" Molecules 26, no. 19: 5788. https://doi.org/10.3390/molecules26195788
APA StylePerera, Y. R., Xu, J. X., Amarasekara, D. L., Hughes, A. C., Abbood, I., & Fitzkee, N. C. (2021). Understanding the Adsorption of Peptides and Proteins onto PEGylated Gold Nanoparticles. Molecules, 26(19), 5788. https://doi.org/10.3390/molecules26195788