
Computational Screening of Natural Compounds for Identification of Potential Anti-Cancer Agents Targeting MCM7 Protein

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Methodology
2.1. Protein Preparation
2.2. Compound Library Preparation
2.3. Receptor-Based Virtual Screening
2.4. Molecular Docking
2.5. Pharmacokinetics and Toxicity Prediction
2.6. Molecular Dynamics Simulations
3. Result and Discussion
3.1. Virtual Screening and Molecular Docking
3.2. Physicochemical and Drug Likeness Properties
3.3. MD Simulations
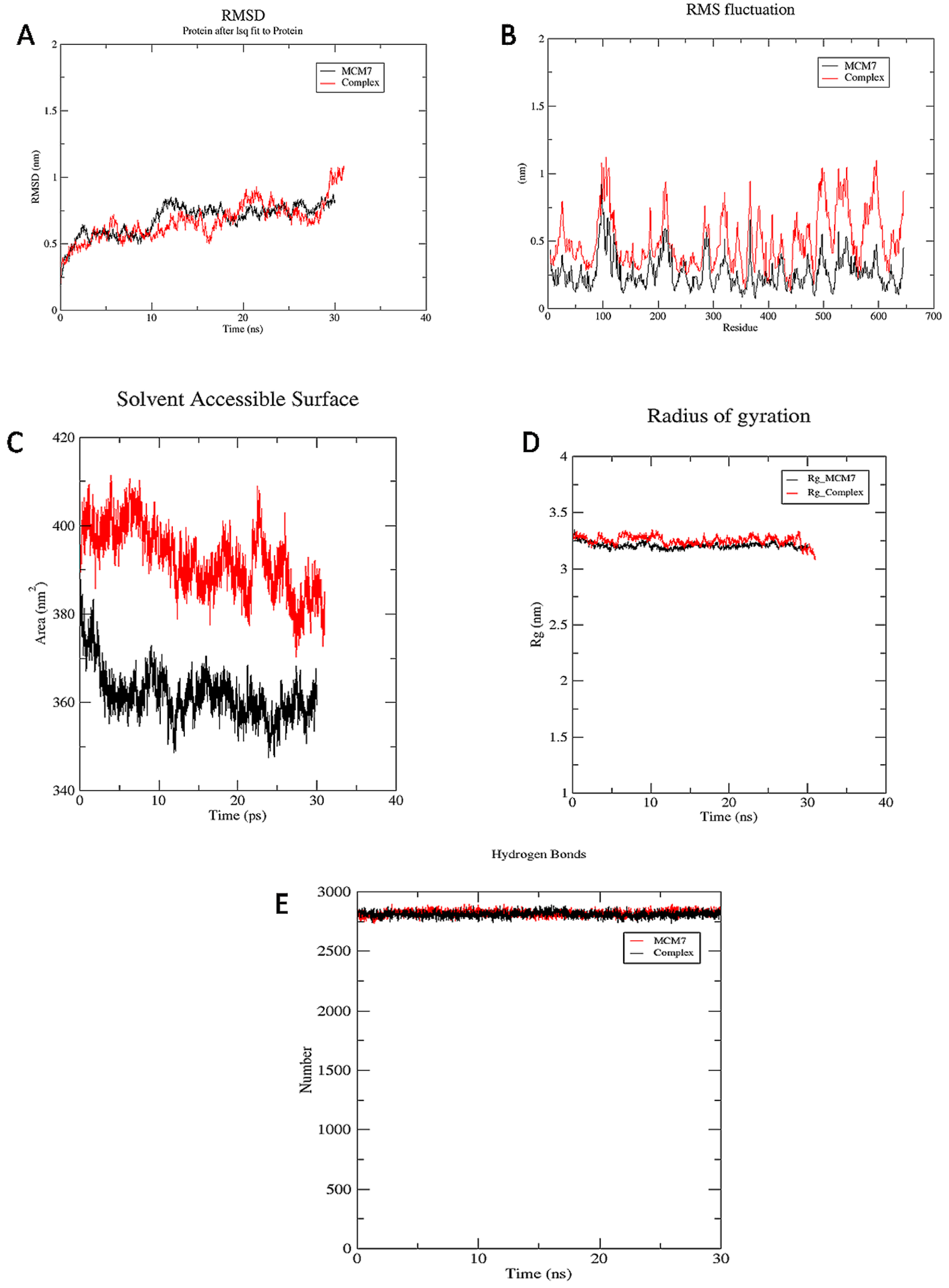
3.3.1. RMSD
3.3.2. RMSF
3.3.3. SASA
3.3.4. Rg
3.3.5. Hydrogen Bond
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Vassilev, A.; DePamphilis, M.L. Links between DNA replication, stem cells and cancer. Genes 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochman, M.L.; Schwacha, A. The Mcm complex: Unwinding the mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. 2009, 73, 652–683. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.G.; Sanders, A.J.; Katoh, M.; Ungefroren, H.; Gieseler, F.; Prince, M.; Thompson, S.; Zollo, M.; Spano, D.; Dhawan, P. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin. Cancer Biol. 2015, 35, S244–S275. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Moll, U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat. Rev. Drug Discov. 2014, 13, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Ye, L.; Sanders, A.J.; Lane, J.; Jiang, W.G. Cancer invasion and metastasis: Molecular and cellular perspective. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Honeycutt, K.A.; Chen, Z.; Koster, M.I.; Miers, M.; Nuchtern, J.; Hicks, J.; Roop, D.R.; Shohet, J.M. Deregulated minichromosomal maintenance protein MCM7 contributes to oncogene driven tumorigenesis. Oncogene 2006, 25, 4027–4032. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.M.; Chovatiya, R.J.; Jameson, N.E.; Turner, D.M.; Zhu, G.; Werner, S.; Huryn, D.M.; Pipas, J.M.; Day, B.W.; Wipf, P.; et al. Pyrimidinone-peptoid hybrid molecules with distinct effects on molecular chaperone function and cell proliferation. Bioorg. Med. Chem. 2008, 16, 3291–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Brosh, R.M., Jr. New insights into DNA helicases as druggable targets for cancer therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef]
- Simon, N.; Bochman, M.L.; Seguin, S.; Brodsky, J.L.; Seibel, W.L.; Schwacha, A. Ciprofloxacin is an inhibitor of the Mcm2-7 replicative helicase. Biosci. Rep. 2013, 33, 00072. [Google Scholar] [CrossRef]
- Ishimi, Y.; Sugiyama, T.; Nakaya, R.; Kanamori, M.; Kohno, T.; Enomoto, T.; Chino, M. Effect of heliquinomycin on the activity of human minichromosome maintenance 4/6/7 helicase. FEBS J. 2009, 276, 3382–3391. [Google Scholar] [CrossRef]
- Toyokawa, G.; Masuda, K.; Daigo, Y.; Cho, H.S.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Chino, M.; Field, H.I.; et al. Minichromosome Maintenance Protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-small cell lung cancer. Mol. Cancer 2011, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- Vignais, P.V.; Lunardi, J. Chemical probes of the mitochondrial ATP synthesis and translocation. Annu. Rev. Biochem. 1985, 54, 977–1014. [Google Scholar] [CrossRef]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharm. 2019, 10, 1614. [Google Scholar] [CrossRef] [Green Version]
- Kinch, M.S. An analysis of FDA-approved drugs for oncology. Drug Discov. Today 2014, 19, 1831–1835. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Analysis of FDA approved anticancer drugs reveals the future of cancer therapy. Cell Cycle 2004, 3, 1035–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, G.C.; Barle, E.L.; Galati, G.; Kluwe, W.M. Functional differentiation of cytotoxic cancer drugs and targeted cancer therapeutics. Regul. Toxicol. Pharmacol. RTP 2014, 70, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.B.; Cavenee, W.K. A tale of two approaches: Complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.H.; He, B. Molecular markers as therapeutic targets in lung cancer. Chin. J. cancer 2013, 32, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.A.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef]
- Langdon, S.R.; Westwood, I.M.; van Montfort, R.L.; Brown, N.; Blagg, J. Scaffold-focused virtual screening: Prospective application to the discovery of TTK inhibitors. J. Chem. Inf. Model. 2013, 53, 1100–1112. [Google Scholar] [CrossRef]
- Rzechorzek, N.J.; Hardwick, S.W.; Jatikusumo, V.A.; Chirgadze, D.Y.; Pellegrini, L. CryoEM structures of human CMG-ATPgammaS-DNA and CMG-AND-1 complexes. Nucleic Acids Res. 2020, 48, 6980–6995. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Li, D.; Zhang, Y.; Guan, B.X.; Gao, P.; Zhou, X.C.; Zhou, C.J. The Expression of MCM7 is a Useful Biomarker in the Early Diagnostic of Gastric Cancer. Pathol. Oncol. Res. 2018, 24, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Clayton, A.C.; Minot, D.M.; Shridhar, V.; Hartmann, L.C.; Gilks, C.B.; Chien, J.R. Minichromosome maintenance protein 7 as a potential prognostic factor for progression-free survival in high-grade serous carcinomas of the ovary. Mod. Pathol. 2011, 24, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Chen, X.; Guan, X.; Zhang, H.; Ma, Y.; Zhang, S.; Wang, E.; Zhang, L.; Han, Y. Overexpression of G9a and MCM7 in oesophageal squamous cell carcinoma is associated with poor prognosis. Histopathology 2015, 66, 192–200. [Google Scholar] [CrossRef]
- Ko, J.; Murga, L.F.; Wei, Y.; Ondrechen, M.J. Prediction of active sites for protein structures from computed chemical properties. Bioinformatics 2005, 21, 258–265. [Google Scholar] [CrossRef]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, 1501240. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Rocha, A.B.; Lopes, R.M.; Schwartsmann, G. Natural products in anticancer therapy. Curr. Opin. Pharmacol. 2001, 1, 364–369. [Google Scholar] [CrossRef]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Lien, J.C.; Huang, L.J.; Teng, C.M.; Wang, J.P.; Kuo, S.C. Synthesis of 2-alkoxy 1,4-naphthoquinone derivatives as antiplatelet, antiinflammatory, and antiallergic agents. Chem. Pharm. Bull. 2002, 50, 672–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.T.; Kuo, H.S.; Hsiao, C.L.; Lin, Y.L. Efficient synthesis of ‘redox-switched’ naphthoquinone thiol-crown ethers and their biological activity evaluation. Bioorg. Med. Chem. 2002, 10, 1947–1952. [Google Scholar] [CrossRef]
- Huang, L.J.; Chang, F.C.; Lee, K.H.; Wang, J.P.; Teng, C.M.; Kuo, S.C. Synthesis and antiplatelet, antiinflammatory, and antiallergic activities of substituted 3-chloro-5,8-dimethoxy-1,4-naphthoquinone and related compounds. Bioorg. Med. Chem. 1998, 6, 2261–2269. [Google Scholar] [CrossRef]
- Jin, Y.R.; Ryu, C.K.; Moon, C.K.; Cho, M.R.; Yun, Y.P. Inhibitory effects of J78, a newly synthesized 1,4-naphthoquinone derivative, on experimental thrombosis and platelet aggregation. Pharmacology 2004, 70, 195–200. [Google Scholar] [CrossRef]
- Coates, A.; Abraham, S.; Kaye, S.B.; Sowerbutts, T.; Frewin, C.; Fox, R.M.; Tattersall, M.H. On the receiving end--patient perception of the side-effects of cancer chemotherapy. Eur. J. Cancer Clin. Oncol. 1983, 19, 203–208. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Clarke, M.; Collins, R.; Darby, S.; Davies, C.; Elphinstone, P.; Evans, V.; Godwin, J.; Gray, R.; Hicks, C.; James, S.; et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 366, 2087–2106. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Zainab, T.; Shakil, S.; Rizvi, S.M. A neuroinformatics study to compare inhibition efficiency of three natural ligands (Fawcettimine, Cernuine and Lycodine) against human brain acetylcholinesterase. Network 2015, 26, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Rizvi, S.M.; Hameed, N.; Biswas, D.; Khan, M.; Shakil, S.; Kamal, M.A. Aptiom (eslicarbazepine acetate) as a dual inhibitor of beta-secretase and voltage-gated sodium channel: Advancement in Alzheimer’s disease-epilepsy linkage via an enzoinformatics study. CNS Neurol. Disord. Drug Targets 2014, 13, 1258–1262. [Google Scholar] [CrossRef]
- Copeland, R.A. Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 2011, 3, 1491–1501. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem. Med. Chem. 2016, 11, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compounds | 2D Structure | Binding Energy (kcal/mol) | Inhibition Constant (µM) | Interacting Residues |
|---|---|---|---|---|---|
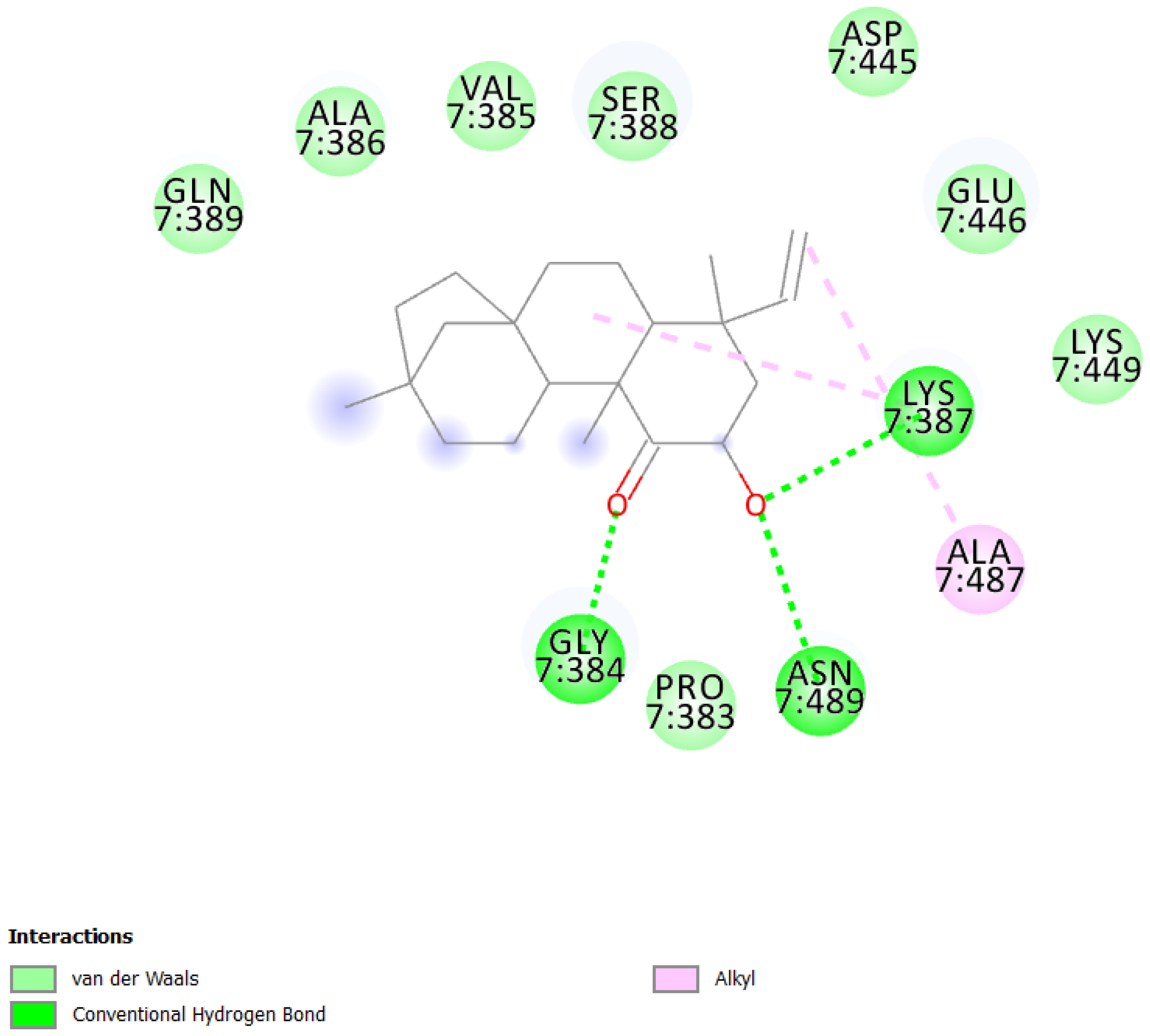
| 1 | UEFS99 |  | −9.95 | 2.49 | Pro383, Gly384, Val385, Ala386, Lys387, Ser388, Gln389, Asp445, Glu446, Lys449, Ala487, and Asn489 |
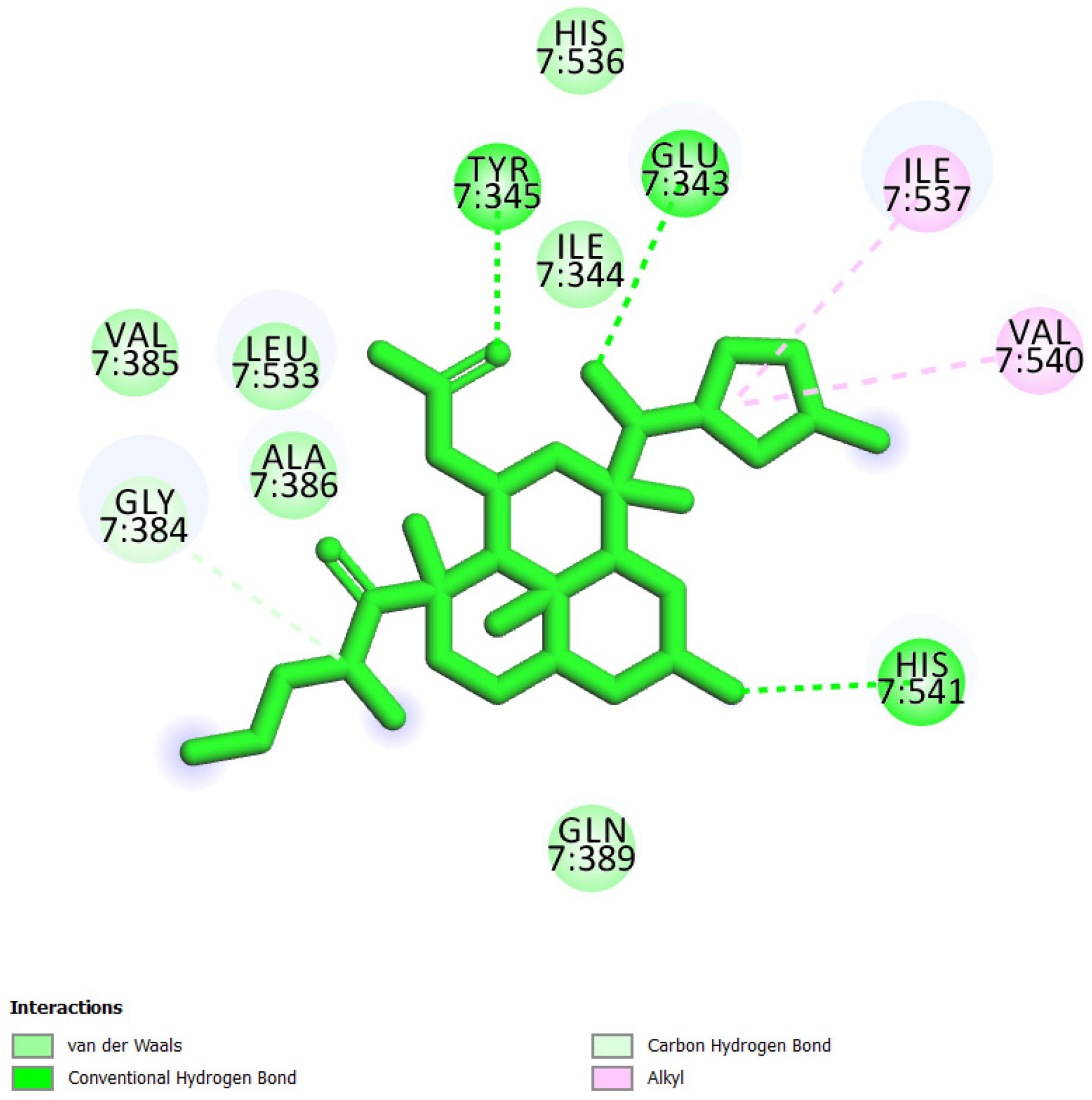
| 2 | UEFS137 |  | −8.92 | 5.13 | Glu343, Ile344, Tyr345, Gly384, Val385, Ala386, Gln389, Leu533, Ile537, His536, Val540, and His541 |
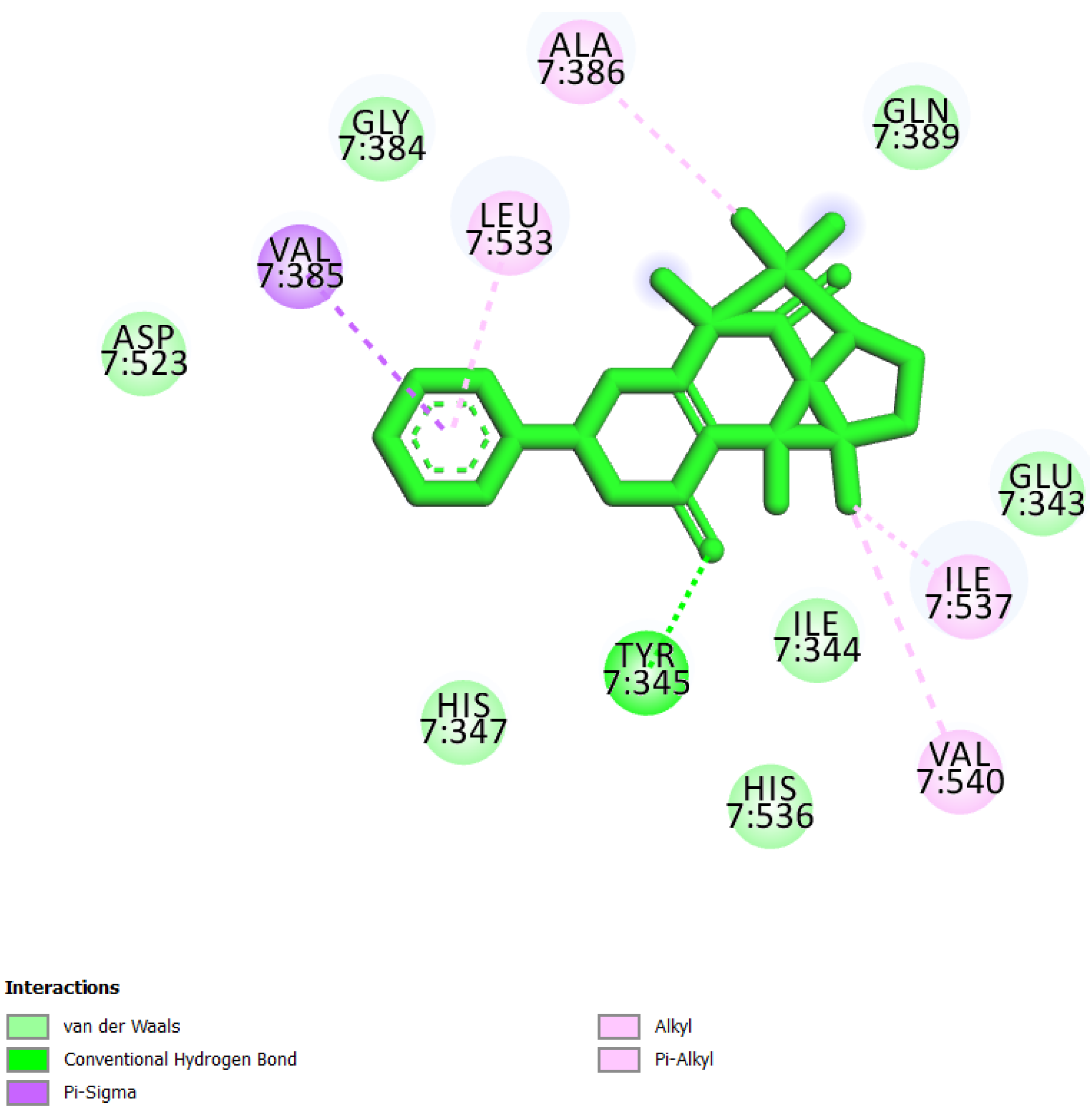
| 3 | UEFS428 |  | −8.71 | 9.53 | Glu343, Ile344, Tyr345, His347, Gly384, Val385, Ala386, Gln389, Asp523, Leu533, His536, Ile537, and Val540 |
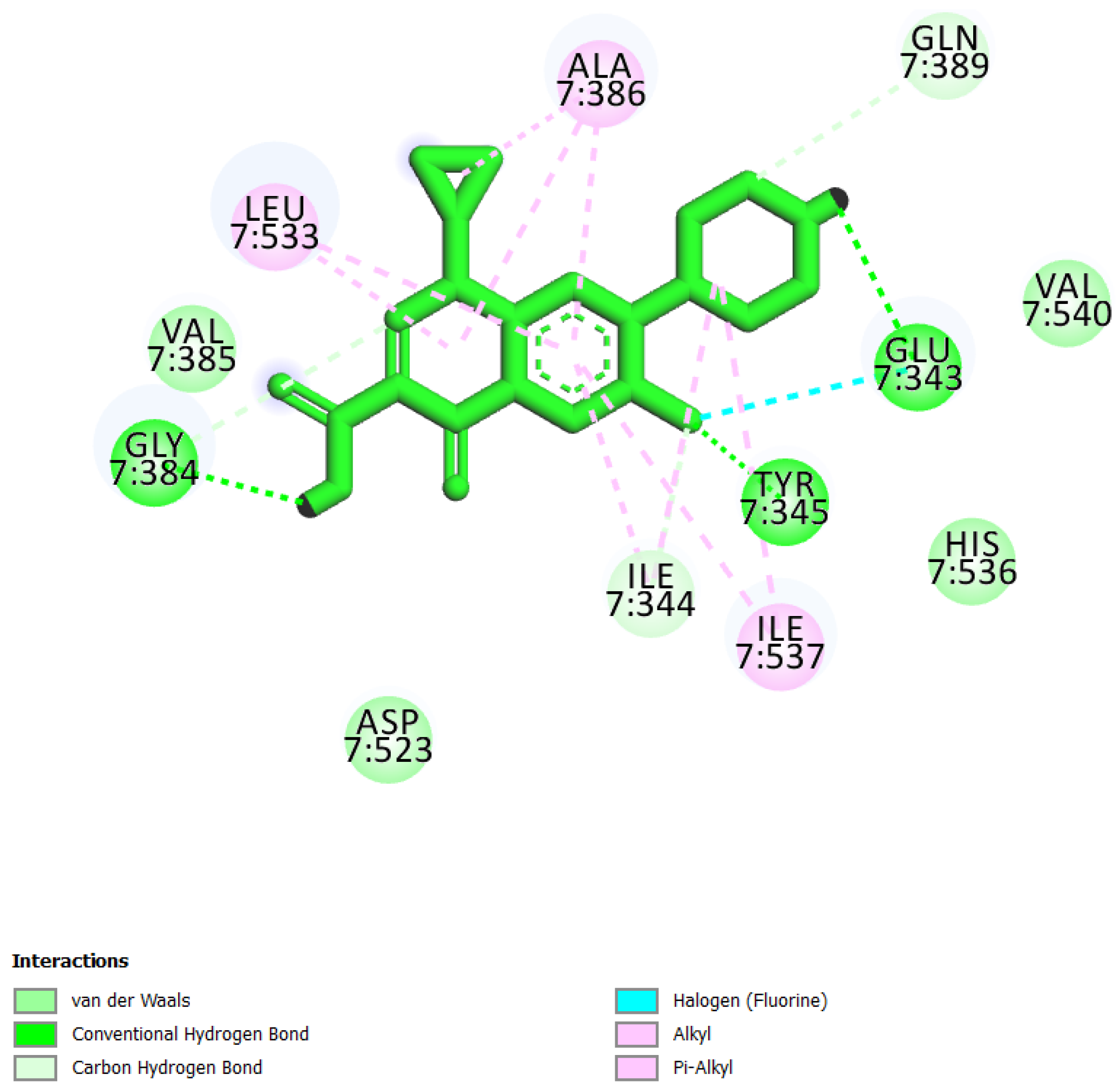
| 4 | Ciprofloxacin * |  | −6.50 | 52.27 | Glu343, Ile344, Tyr345, Gly384, Val385, Ala386, Gln389, Asp523, Leu533, Ile537, and Val540 |
| Property | Model Name | Predicted Value | |||
|---|---|---|---|---|---|
| UEFS 428 | UEFS 137 | UEFS 99 | |||
 |  |  | |||
| Physicochemical Properties | MW | 380.43 | 504.57 | 316.48 | |
| MR | 100.3 | 125.97 | 94.45 | ||
| TPSA | 72.83 | 133.27 | 37.3 | ||
| Lipophilicity | iLOGP | 0 | 1.6 | 2.99 | |
| XLOGP3 | 1.02 | 1.59 | 5.67 | ||
| WLOGP | 2.41 | 2.01 | 4.52 | ||
| MLOGP | 1.56 | 1.73 | 4.05 | ||
| Silicos-IT Log P | 4.04 | 2.87 | 4.3 | ||
| Consensus Log P | 1.81 | 1.96 | 4.3 | ||
| Estimated SOLubility (ESOL) | Log S | −2.93 | −3.71 | −5.31 | |
| Solubility (mg/mL) | 4.43 × 10−1 | 9.93 × 10−2 | 1.56 × 10−3 | ||
| Solubility (mol/L) | 1.16 × 10−3 | 1.97 × 10−4 | 4.92 × 10−6 | ||
| Class | Soluble | Soluble | Moderately soluble | ||
| Pharmacokinetics | GI absorption | High | High | High | |
| BBB permeant | Yes | No | Yes | ||
| Pgp substrate | Yes | No | |||
| inhibitor | CYP1A2 | No | No | No | |
| CYP2C19 | |||||
| CYP2C9 | Yes | ||||
| CYP2D6 | No | ||||
| CYP3A4 | |||||
| log Kp (cm/s) | −7.9 | −8.25 | −4.2 | ||
| Druglikeness | Lipinski | Number of violations | 0 | 1 | 0 |
| Ghose | 0 | 2 | 0 | ||
| Veber | 0 | 0 | 0 | ||
| Egan | 0 | 1 | 0 | ||
| Muegge | 0 | 0 | 1 | ||
| Classification | Target | UEFS 428 | UEFS 137 | UEFS 99 | |||
|---|---|---|---|---|---|---|---|
| Prediction | Probability | Prediction | Probability | Prediction | Probability | ||
| Organ toxicity | Hepatotoxicity | - | 0.75 | - | 0.85 | - | 0.71 |
| Toxicity end points | Carcinogenicity | Active | 0.51 | - | 0.61 | - | 0.67 |
| Immunotoxicity | - | 0.96 | Active | 0.99 | Active | 0.97 | |
| Mutagenicity | - | 0.52 | - | 0.74 | - | 0.86 | |
| Cytotoxicity | - | 0.61 | - | 0.76 | - | 0.76 | |
| Tox21-Nuclear receptor signalling pathways | Aryl hydrocarbon Receptor | - | 0.9 | - | 0.97 | - | 0.98 |
| Androgen Receptor | - | 0.83 | - | 0.84 | - | 0.52 | |
| Androgen Receptor Ligand Binding Domain | - | 0.9 | - | 0.88 | - | 0.52 | |
| Aromatase | - | 0.84 | - | 0.84 | - | 0.96 | |
| Estrogen Receptor Alpha | - | 0.66 | - | 0.73 | Active | 0.53 | |
| Estrogen Receptor Ligand Binding Domain | - | 0.98 | - | 0.95 | - | 0.61 | |
| PPAR-Gamma | - | 0.93 | - | 0.92 | - | 0.98 | |
| Tox21-Stress response pathways | nrf2/ARE | - | 0.87 | - | 0.98 | - | 0.91 |
| Heat shock factor response element | - | 0.87 | - | 0.98 | - | 0.91 | |
| Mitochondrial Membrane Potential | - | 0.57 | - | 0.72 | - | 0.74 | |
| p53 | - | 0.8 | - | 0.83 | - | 0.94 | |
| ATPase family AAA domain-containing protein 5 | - | 0.9 | - | 0.91 | - | 0.96 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshahrani, M.Y.; Alshahrani, K.M.; Tasleem, M.; Akeel, A.; Almeleebia, T.M.; Ahmad, I.; Asiri, M.; Alshahrani, N.A.; Alabdallah, N.M.; Saeed, M. Computational Screening of Natural Compounds for Identification of Potential Anti-Cancer Agents Targeting MCM7 Protein. Molecules 2021, 26, 5878. https://doi.org/10.3390/molecules26195878
Alshahrani MY, Alshahrani KM, Tasleem M, Akeel A, Almeleebia TM, Ahmad I, Asiri M, Alshahrani NA, Alabdallah NM, Saeed M. Computational Screening of Natural Compounds for Identification of Potential Anti-Cancer Agents Targeting MCM7 Protein. Molecules. 2021; 26(19):5878. https://doi.org/10.3390/molecules26195878
Chicago/Turabian StyleAlshahrani, Mohammad Y., Kholoud M. Alshahrani, Munazzah Tasleem, Arshiya Akeel, Tahani M. Almeleebia, Irfan Ahmad, Mohammed Asiri, Najla A. Alshahrani, Nadiyah M. Alabdallah, and Mohd Saeed. 2021. "Computational Screening of Natural Compounds for Identification of Potential Anti-Cancer Agents Targeting MCM7 Protein" Molecules 26, no. 19: 5878. https://doi.org/10.3390/molecules26195878
APA StyleAlshahrani, M. Y., Alshahrani, K. M., Tasleem, M., Akeel, A., Almeleebia, T. M., Ahmad, I., Asiri, M., Alshahrani, N. A., Alabdallah, N. M., & Saeed, M. (2021). Computational Screening of Natural Compounds for Identification of Potential Anti-Cancer Agents Targeting MCM7 Protein. Molecules, 26(19), 5878. https://doi.org/10.3390/molecules26195878