
The Synthesis of Blood Group Antigenic A Trisaccharide and Its Biotinylated Derivative


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
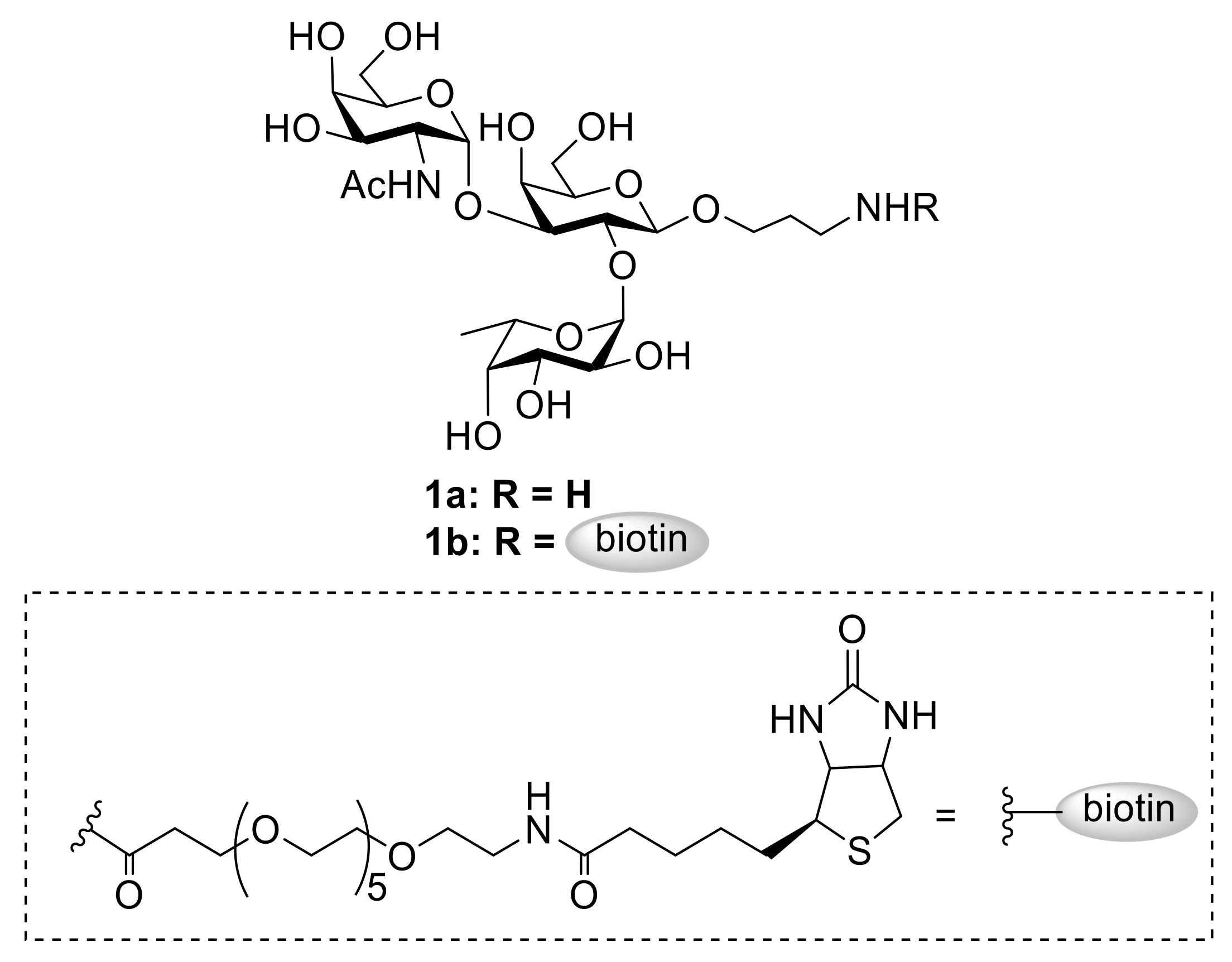
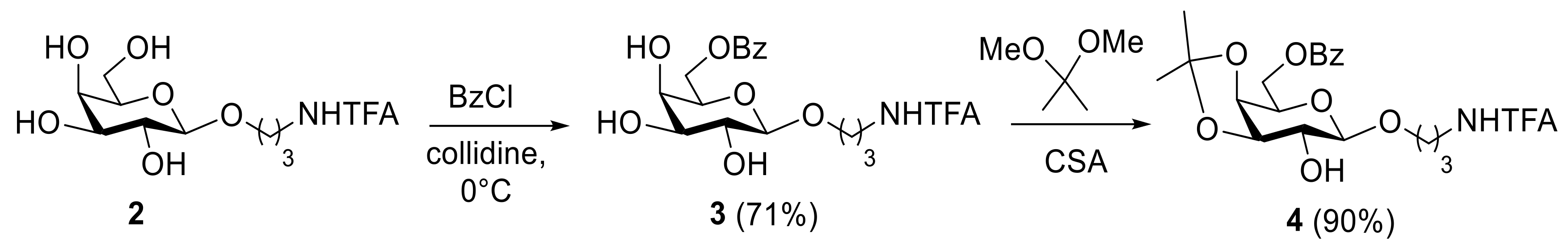
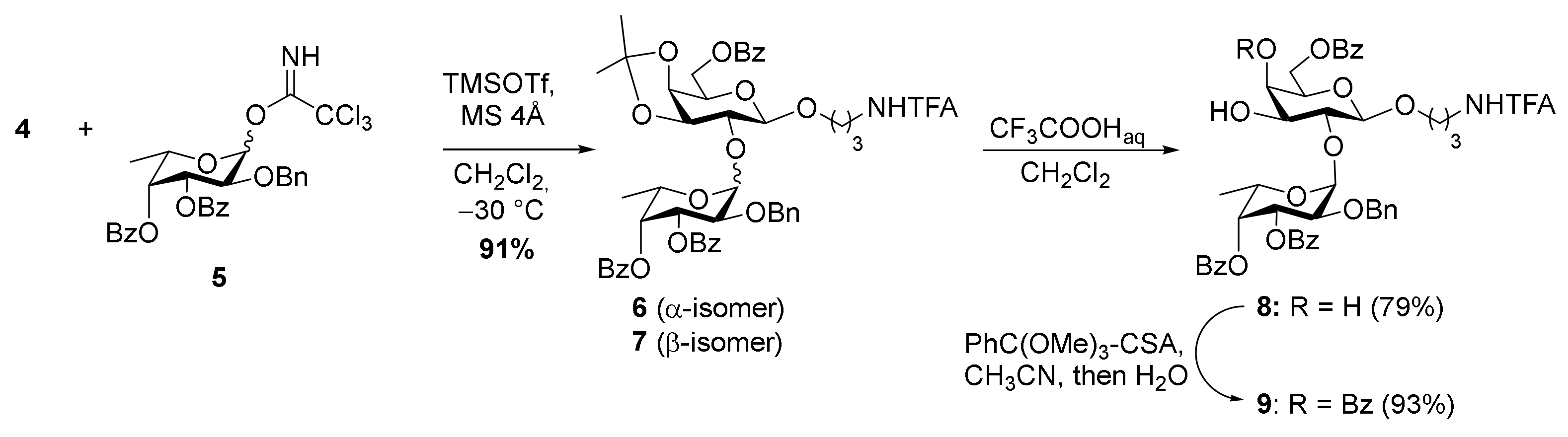
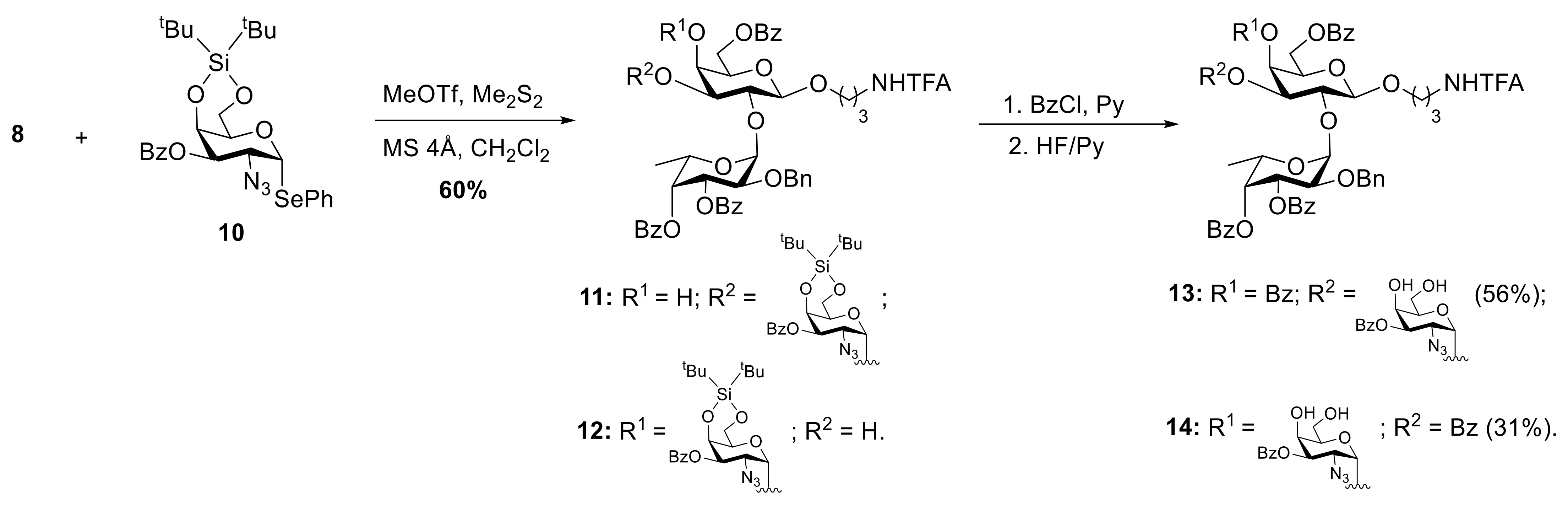
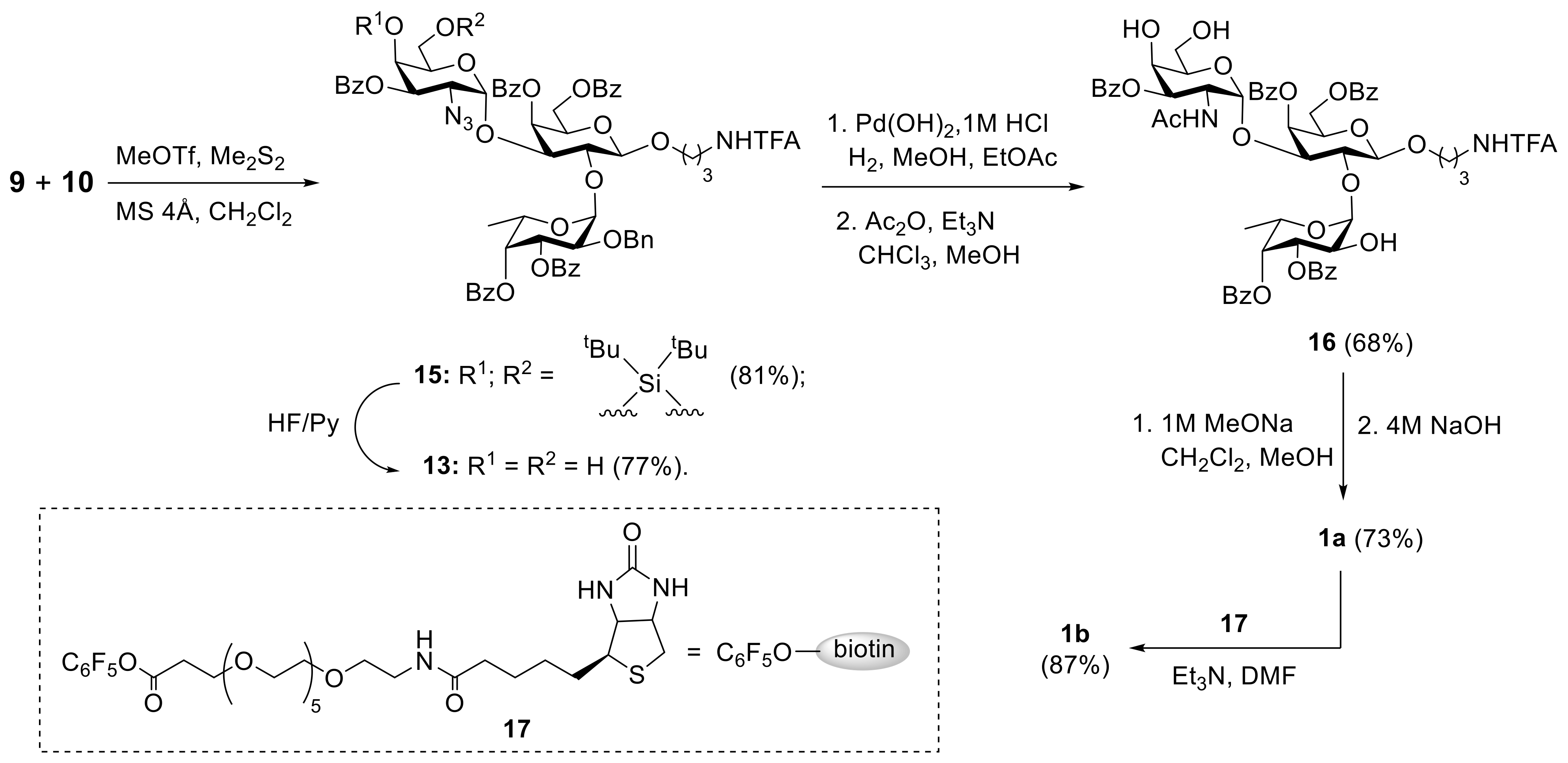
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Compounds 4, 8, 9, 13, 14, 1a and 1b
3.2.1. 3-Trifluoroacetamidopropyl 3,4-O-Isopropylidene-6-O-Benzoyl-β-D-Galactopyranoside (4)
3.2.2. 3-Trifluoroacetamidopropyl 2-O-Benzyl-3,4-Di-O-Benzoyl-α-L-Fucopyranosyl- (1→2)-6-O-Benzoyl-β-D-Galactopyranoside (8)
3.2.3. 3-Trifluoroacetamidopropyl 2-O-Benzyl-3,4-di-O-Benzoyl-α-L-Fucopyranosyl- (1→2)-4,6-Di-O-Benzoyl-β-D-Galactopyranoside (9)
3.2.4. 3-Trifluoroacetamidopropyl 2-Azido-3-O-Benzoyl-2-Deoxy-α-D-Galactopyranosyl- (1→3)-[2-O-Benzyl-3,4-Di-O-Benzoyl-α-L-Fucopyranosyl-(1→2)]-4,6-Di-O-Benzoyl-β-D-Galactopyranoside (13) and 3-Trifluoroacetamidopropyl 2-Azido-3-O-Benzoyl-2-Deoxy-α-D-Galactopyranosyl-(1→4)-[2-O-Benzyl-3,4-Di-O-Benzoyl-α-L-Fucopyranosyl-(1→2)]- 4,6-Di-O-Benzoyl-β-D-Galactopyranoside (14)
3.2.5. 3-Aminopropyl 2-Acetamido-2-Deoxy-α-D-Galactopyranosyl-(l→3)-[(α-L-Fucopyranosyl)-(l→2)]-β-D-Galactopyranoside (1a)
3.2.6. 3-(21-Biotinamino-4,7,10,13,16,19-Hexaoxagenicaminoamino)-Propyl 2-Acetamido-2-Deoxy-α-D-Galactopyranosyl-(l→3)-[(α-L-Fucopyranosyl)-(l→2)]-β-D-Galactopyranoside (1b)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Morgan, W.T.J.; Watkins, W.M. Genetic and Biochemical Aspects of Human Blood-Group A-, B-, H-, Le-a- and Le-b-Specificity. Br. Med. Bull. 1969, 25, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Rege, V.P.; Painter, T.J.; Watkins, W.M.; Morgan, W.T.J. Isolation of Serologically Active Fucose-Containing Oligosaccharides from Human Blood-Group H Substance. Nature 1964, 203, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Bovin, N.V.; Zurabyan, S.É.; Khorlin, A.Y. Stereoselectivity in Glycosylation by Means of 2-Azido-2-Desoxy-D-Galactopyranose Derivatives and the Synthesis of the Determinative Oligosaccharide of Blood Group A, Type 1. Russ. Chem. Bull. 1982, 31, 1023–1030. [Google Scholar] [CrossRef]
- Åberg, P.-M.; Blomberg, L.; Lönn, H.; Norberg, T. Glycosylation with Thioglycosides Activated by Dimethyl(Methylthio)Sulfonium Tetrafluoroborate: Synthesis of Two Trisaccharide Glycosides Corresponding to the Blood Group A and B Determinants. Glycoconj. J. 1990, 7, 201–205. [Google Scholar] [CrossRef]
- EIu, K.; Bovina, N.V. Synthesis of spaced trisaccharides with blood group A and B specificity, their fragments, and structural analogs. Bioorg. Khim. 1992, 18, 283–298. [Google Scholar]
- Khatuntseva, E.A.; Tsvetkov, Y.E.; Grachev, A.A.; Nifant’ev, N.E. Synthesis of Aminoethyl Glycosides of Type 2 Chain A Tetrasaccharide and Related Trisaccharides. Russ. J. Org. Chem. 2005, 41, 1814–1823. [Google Scholar] [CrossRef]
- Meloncelli, P.J.; Lowary, T.L. Synthesis of ABO Histo-Blood Group Type I and II Antigens. Carbohydr. Res. 2010, 345, 2305–2322. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. A New Chemical Approach to Human ABO Histo-Blood Group Type 2 Antigens. Molecules 2014, 19, 414–437. [Google Scholar] [CrossRef] [Green Version]
- Karki, G.; Mishra, V.N.; Mandal, P.K. An Expeditious Synthesis of Blood-Group Antigens, ABO Histo-Blood Group Type II Antigens and Xenoantigen Oligosaccharides with Amino Type Spacer−arms. Glycoconj. J. 2016, 33, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Kunetskiy, R.A.; Pazynina, G.V.; Ivanov, I.A.; Bovin, N.V. Synthesis of Blood Group A and B (Type 2) Tetrasaccharides. A Strategy with Fucosylation at the Last Stage. Carbohydr. Res. 2020, 498, 108192. [Google Scholar] [CrossRef]
- West, L.J.; Pollock-Barziv, S.M.; Dipchand, A.I.; Lee, K.J.; Cardella, C.J.; Benson, L.N.; Rebeyka, I.M.; Coles, J.G. ABO-Incompatible Heart Transplantation in Infants. N. Engl. J. Med. 2001, 344, 793–800. [Google Scholar] [CrossRef]
- Yamamoto, F. Review: ABO Blood Group System--ABH Oligosaccharide Antigens, Anti-A and Anti-B, A and B Glycosyltransferases, and ABO Genes. Immunohematology 2004, 20, 3–22. [Google Scholar] [CrossRef]
- Bentall, A.; Jeyakanthan, M.; Braitch, M.; Cairo, C.W.; Lowary, T.L.; Maier, S.; Halpin, A.; Motyka, B.; Zou, L.; West, L.J.; et al. Characterization of ABH-Subtype Donor-Specific Antibodies in ABO-A-Incompatible Kidney Transplantation. Am. J. Transplant. 2021. [Google Scholar] [CrossRef]
- Casset, F.; Peters, T.; Etzler, M.; Korchagina, E.; Nifant’ev, N.; Pérez, S.; Imberty, A. Conformational Analysis of Blood Group A Trisaccharide in Solution and in the Binding Site of Dolichos Biflorus Lectin Using Transient and Transferred Nuclear Overhauser Enhancement (NOE) and Rotating-Frame NOE Experiments. Eur. J. Biochem. 1996, 239, 710–719. [Google Scholar] [CrossRef]
- Iurisci, I.; Cumashi, A.; Sherman, A.A.; Tsvetkov, Y.E.; Tinari, N.; Piccolo, E.; D’egidio, M.; Adamo, V.; Natoli, C.; Rabinovich, G.A.; et al. Synthetic Inhibitors of Galectin-1 and -3 Selectively Modulate Homotypic Cell Aggregation and Tumor Cell Apoptosis. Anticancer Res. 2009, 29, 403–410. [Google Scholar]
- Quintana, J.I.; Delgado, S.; Núñez-Franco, R.; Cañada, F.J.; Jiménez-Osés, G.; Jiménez-Barbero, J.; Ardá, A. Galectin-4 N-Terminal Domain: Binding Preferences Toward A and B Antigens With Different Peripheral Core Presentations. Front. Chem. 2021, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Lipkind, G.M.; Shashkov, A.S.; Nifant’ev, N.E.; Kochetkov, N.K. Computer-Assisted Analysis of the Structure of Regular Branched Polysaccharides Containing 2,3-Disubstituted Rhamnopyranose and Mannopyranose Residues on the Basis of 13C NMR Data. Carbohydr. Res. 1992, 237, 11–22. [Google Scholar] [CrossRef]
- Gerbst, A.G.; Ustuzhanina, N.E.; Grachev, A.A.; Khatuntseva, E.A.; Tsvetkov, D.E.; Whitfield, D.M.; Berces, A.; Nifantiev, N.E. Synthesis, NMR, and Conformational Studies of Fucoidan Fragments. III. Effect of Benzoyl Group at O-3 on Stereoselectivity of Glycosylation by 3-O- and 3,4-Di-O-Benzoylated 2-O-Benzylfucosyl Bromides. J. Carbohydr. Chem. 2001, 20, 821–831. [Google Scholar] [CrossRef]
- Khatuntseva, E.A.; Ustuzhanina, N.E.; Zatonskii, G.V.; Shashkov, A.S.; Usov, A.I.; Nifantiev, N.E. The Synthesis and NMR and Conformational Studies of Fucoidan Fragments: I Synthesis, NMR and conformational studies of fucoidan fragments. Part 1. Desulfated 2,3- and 3,4-branched trisaccharide fragments and costituing disaccharides. J. Carbohydr. Chem. 2000, 19, 1151–1173. [Google Scholar] [CrossRef]
- Kazakova, E.D.; Yashunsky, D.V.; Krylov, V.B.; Bouchara, J.-P.; Cornet, M.; Valsecchi, I.; Fontaine, T.; Latgé, J.-P.; Nifantiev, N.E. Biotinylated Oligo-α-(1 → 4)-d-Galactosamines and Their N-Acetylated Derivatives: α-Stereoselective Synthesis and Immunology Application. J. Am. Chem. Soc. 2020, 142, 1175–1179. [Google Scholar] [CrossRef] [PubMed]
- Imamura, A.; Matsuzawa, N.; Sakai, S.; Udagawa, T.; Nakashima, S.; Ando, H.; Ishida, H.; Kiso, M. The Origin of High Stereoselectivity in Di-Tert-Butylsilylene-Directed α-Galactosylation. J. Org. Chem. 2016, 81, 9086–9104. [Google Scholar] [CrossRef]
- Komarova, B.S.; Tsvetkov, Y.E.; Nifantiev, N.E. Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chem. Rec. 2016, 16, 488–506. [Google Scholar] [CrossRef]
- Kazakova, E.D.; Yashunsky, D.V.; Khatuntseva, E.A.; Nifantiev, N.E. Azidophenylselenylation of Glycals towards 2-Azido-2-Deoxy-Selenoglycosides and Their Application in Oligosaccharide Synthesis. Pure Appl. Chem. 2020, 92, 1047–1056. [Google Scholar] [CrossRef]
- Nigudkar, S.S.; Demchenko, A.V. Stereocontrolled 1,2- Cis Glycosylation as the Driving Force of Progress in Synthetic Carbohydrate Chemistry. Chem. Sci. 2015, 6, 2687–2704. [Google Scholar] [CrossRef] [Green Version]
- Milner, S.J.; Carrick, C.T.; Kerr, K.G.; Snelling, A.M.; Thomas, G.H.; Duhme-Klair, A.-K.; Routledge, A. Probing Bacterial Uptake of Glycosylated Ciprofloxacin Conjugates. ChemBioChem 2014, 15, 466–471. [Google Scholar] [CrossRef]
- Kochetkov, N.K.; Nifant’ev, N.E.; Backinowsky, L.V. Synthesis of the Capsular Polysaccharide of Streptococcus Pneumoniae Type 14. Tetrahedron 1987, 43, 3109–3121. [Google Scholar] [CrossRef]
- Mironov, Y.V.; Sherman, A.A.; Nifantiev, N.E. Homogeneous Azidophenylselenylation of Glycals Using TMSN3–Ph2Se2–PhI(OAc)2. Tetrahedron Lett. 2004, 45, 9107–9110. [Google Scholar] [CrossRef]
- Khatuntseva, E.A.; Sherman, A.A.; Tsvetkov, Y.E.; Nifantiev, N.E. Phenyl 2-Azido-2-Deoxy-1-Selenogalactosides: A Single Type of Glycosyl Donor for the Highly Stereoselective Synthesis of α- and β-2-Azido-2-Deoxy-d-Galactopyranosides. Tetrahedron Lett. 2016, 57, 708–711. [Google Scholar] [CrossRef]
- Hagen, B.; Ali, S.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Mapping the Reactivity and Selectivity of 2-Azidofucosyl Donors for the Assembly of N-Acetylfucosamine-Containing Bacterial Oligosaccharides. J. Org. Chem. 2017, 82, 848–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gómez-Redondo, M.; Jiménez-Osés, G.; Arda, A.; Overkleeft, H.S.; van der Marel, G.A.; Jiménez-Barbero, J.; Codée, J.D.C. Synthesis and Structural Analysis of Aspergillus Fumigatus Galactosaminogalactans Featuring α-Galactose, α-Galactosamine and α-N-Acetyl Galactosamine Linkages. Angew. Chem. Int. Ed. 2020, 59, 12746–12750. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, G.; Du, W.; Zhang, Y.; Xing, G. A Novel O-Fucosylation Strategy Preactivated by (p-Tol)2SO/Tf2O and Its Application for the Synthesis of Lewis Blood Group Antigen Lewisa. Tetrahedron Lett. 2017, 58, 2109–2112. [Google Scholar] [CrossRef]
- Vigo, E.A.D.; Stortz, C.A.; Marino, C. Regioselectivity of Glycosylation Reactions of Galactose Acceptors: An Experimental and Theoretical Study. Beilstein J. Org. Chem. 2019, 15, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, Y.E.; Burg-Roderfeld, M.; Loers, G.; Ardá, A.; Sukhova, E.V.; Khatuntseva, E.A.; Grachev, A.A.; Chizhov, A.O.; Siebert, H.-C.; Schachner, M.; et al. Synthesis and Molecular Recognition Studies of the HNK-1 Trisaccharide and Related Oligosaccharides. The Specificity of Monoclonal Anti-HNK-1 Antibodies as Assessed by Surface Plasmon Resonance and STD NMR. J. Am. Chem. Soc. 2012, 134, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazakova, E.D.; Yashunsky, D.V.; Nifantiev, N.E. The Synthesis of Blood Group Antigenic A Trisaccharide and Its Biotinylated Derivative. Molecules 2021, 26, 5887. https://doi.org/10.3390/molecules26195887
Kazakova ED, Yashunsky DV, Nifantiev NE. The Synthesis of Blood Group Antigenic A Trisaccharide and Its Biotinylated Derivative. Molecules. 2021; 26(19):5887. https://doi.org/10.3390/molecules26195887
Chicago/Turabian StyleKazakova, Ekaterina D., Dmitry V. Yashunsky, and Nikolay E. Nifantiev. 2021. "The Synthesis of Blood Group Antigenic A Trisaccharide and Its Biotinylated Derivative" Molecules 26, no. 19: 5887. https://doi.org/10.3390/molecules26195887
APA StyleKazakova, E. D., Yashunsky, D. V., & Nifantiev, N. E. (2021). The Synthesis of Blood Group Antigenic A Trisaccharide and Its Biotinylated Derivative. Molecules, 26(19), 5887. https://doi.org/10.3390/molecules26195887