
Approaches to the Synthesis of Dicarboxylic Derivatives of Bis(pyrazol-1-yl)alkanes



Abstract
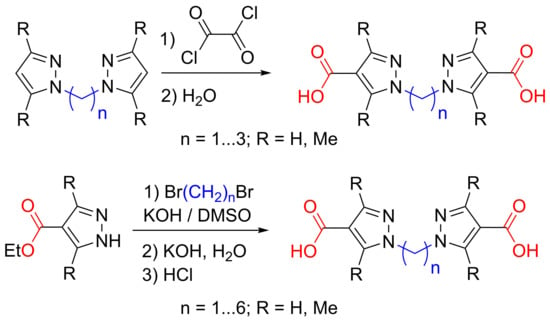
:
1. Introduction
2. Results and Discussion
- (1)
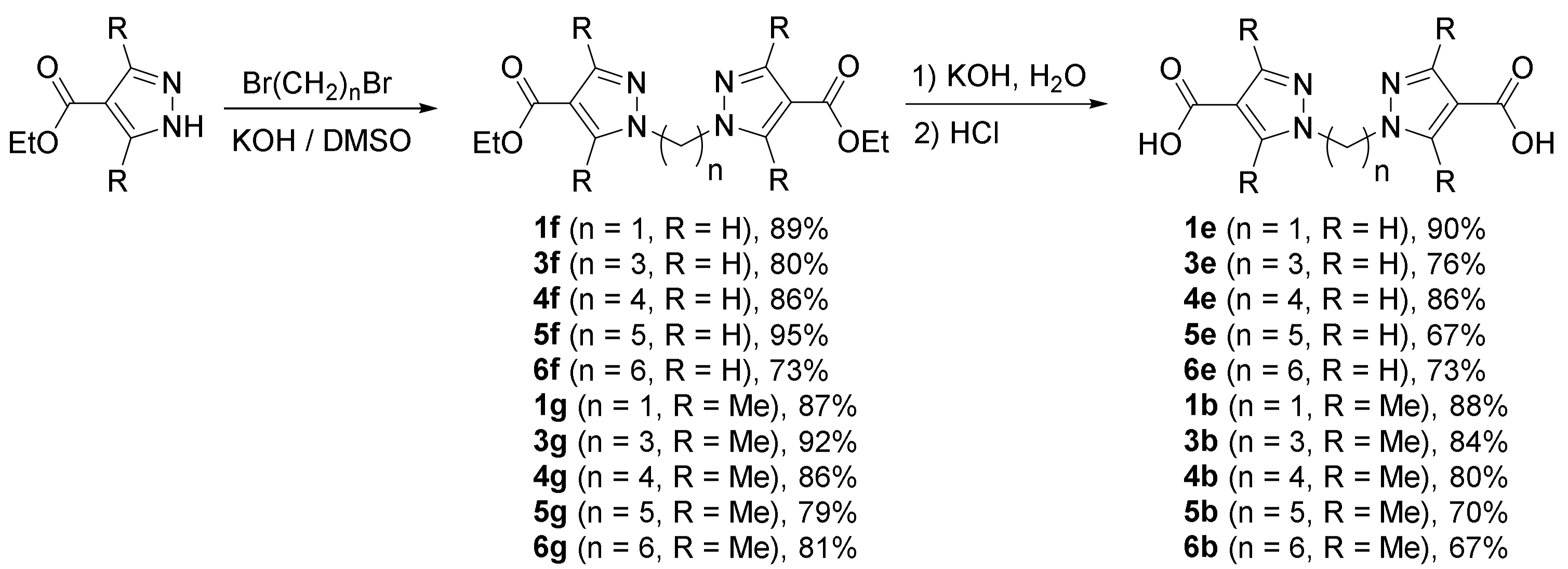
- The introduction of methyl groups into pyrazole rings (in pairs of compounds 1a–1d and 3a–3d) noticeably increases the negative charge at position 4 of the heterocycle, i.e., makes it more active in the electrophilic substitution reaction, and the effect of electron-donor groups is best manifested when comparing the sum of charges on carbon and hydrogen atoms;
- (2)
- An increase in the length of the linker from one to three methylene groups also increases the excess negative charge at position 4, which is apparently associated with the negative inductive effect of the pyrazole ring;
- (3)
- The introduction of an electron-withdrawing acid chloride group into one of the pyrazole rings deactivates the other cycle in the electrophilic substitution reaction, and to a greater extent, deactivation manifests itself in pyrazole derivatives without methyl substituents, and with a short methylene linker.
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pettinari, C.; Pettinari, R. Metal Derivatives of Poly(pyrazolyl)alkanes: II. Bis(pyrazolyl)alkanes and Related Systems. Coord. Chem. Rev. 2005, 249, 663–691. [Google Scholar] [CrossRef]
- Dehury, N.; Tripathy, S.K.; Sahoo, A.; Maity, N.; Patra, S. Facile Tandem Suzuki Coupling/Transfer Hydrogenation Reaction with a Bis-Heteroscorpionate Pd-Ru Complex. Dalton Trans. 2014, 43, 16597–16600. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Otero, A.; Lara-Sánchez, A.; Castro-Osma, J.A.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Rodríguez, A.M. Heteroscorpionate Rare-Earth Catalysts for the Hydroalkoxylation/Cyclization of Alkynyl Alcohols. Organometallics 2016, 35, 1802–1812. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Lara-Sánchez, A.; Sánchez-Barba, L.F. Metal Complexes with Heteroscorpionate Ligands Based on the Bis(pyrazol-1-yl)methane Moiety: Catalytic Chemistry. Coord. Chem. Rev. 2013, 257, 1806–1868. [Google Scholar] [CrossRef]
- Zubkevich, S.V.; Tuskaev, V.A.; Gagieva, S.C.; Pavlov, A.A.; Khrustalev, V.N.; Polyakova, O.V.; Zarubin, D.N.; Kurmaev, D.A.; Kolosov, N.A.; Bulychev, B.M. Catalytic Systems Based on Nickel(ii) Complexes with Bis(3,5-dimethylpyrazol-1-yl)methane—Impact of PPh3 on the Formation of Precatalysts and Selective Dimerization of Ethylene. New J. Chem. 2020, 44, 981–993. [Google Scholar] [CrossRef]
- Moegling, J.; Hoffmann, A.; Herres-Pawlis, S. Insights into Copper-Poly(pyrazolyl)methane-Catalyzed Reactions for Organic Transformations. Synthesis 2017, 49, 225–236. [Google Scholar]
- Montani, M.; Pazmay, G.V.B.; Hysi, A.; Lupidi, G.; Pettinari, R.; Gambini, V.; Tilio, M.; Marchetti, F.; Pettinari, C.; Ferraro, S.; et al. The Water Soluble Ruthenium(II) Organometallic Compound [Ru(p-cymene)(bis(3,5 dimethylpyrazol-1-yl)methane)Cl]Cl Suppresses Triple Negative Breast Cancer Growth by Inhibiting Tumor Infiltration of Regulatory T Cells. Pharmacol. Res. 2016, 107, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, S.; Cirilli, I.; Marcheggiani, F.; Dludla, P.; Lupidi, G.; Pettinari, R.; Marchetti, F.; Di Nicola, C.; Falcioni, G.; Marchini, C.; et al. Evaluation of Anticancer Role of a Novel Ruthenium(II)-Based Compound Compared with NAMI-A and Cisplatin in Impairing Mitochondrial Functionality and Promoting Oxidative Stress in Triple Negative Breast Cancer Models. Mitochondrion 2021, 56, 25–34. [Google Scholar] [CrossRef]
- Fonseca, D.; Páez, C.; Ibarra, L.; García-Huertas, P.; Macías, M.A.; Triana-Chávez, O.; Hurtado, J.J. Metal Complex Derivatives of Bis(pyrazol-1-yl)methane Ligands: Synthesis, Characterization and Anti-Trypanosoma Cruzi Activity. Transit. Met. Chem. 2019, 44, 135–144. [Google Scholar] [CrossRef]
- Schepetkin, I.; Potapov, A.; Khlebnikov, A.; Korotkova, E.; Lukina, A.; Malovichko, G.; Kirpotina, L.; Quinn, M.T. Decomposition of Reactive Oxygen Species by Copper(II) bis(1-pyrazolyl) Methane Complexes. J. Biol. Inorg. Chem. 2006, 11, 499–513. [Google Scholar] [CrossRef]
- Potapov, A.S.; Nudnova, E.A.; Domina, G.A.; Kirpotina, L.N.; Quinn, M.T.; Khlebnikov, A.I.; Schepetkin, I.A. Synthesis, Characterization and Potent Superoxide Dismutase-Like Activity of Novel Bis(pyrazole)-2,2′-bipyridyl Mixed Ligand Copper(II) Complexes. Dalton Trans. 2009, 4488–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Li, W.; Wei, D.; Li, C.; Pan, C.; Dong, X.; Li, Z.; Li, S.; Wei, B.; Zhang, F.; et al. Synthesis, Characterization, Photo- and Electro-Luminescent Properties of Blue Cationic Iridium Complexes with Nonconjugated Bis(pyrazole-1-yl)methane as the Ancillary Ligand. Dye. Pigment. 2016, 134, 19–26. [Google Scholar] [CrossRef]
- Meng, S.; Jung, I.; Feng, J.; Scopelliti, R.; Di Censo, D.; Grätzel, M.; Nazeeruddin, M.K.; Baranoff, E. Bis(pyrazol-1-yl)methane as Non-Chromophoric Ancillary Ligand for Charged Bis-Cyclometalated Iridium(III) Complexes. Eur. J. Inorg. Chem. 2012, 2012, 3209–3215. [Google Scholar] [CrossRef]
- Alkorta, I.; Claramunt, R.M.; Díez-Barra, E.; Elguero, J.; de la Hoz, A.; López, C. The Organic Chemistry of Poly(1H-pyrazol-1-yl)methanes. Coord. Chem. Rev. 2017, 339, 153–182. [Google Scholar] [CrossRef]
- Potapov, A.S.; Khlebnikov, A.I.; Vasilevskii, S.F. Synthesis of Monomeric and Oligomeric 1,1′-methylenebis-(1H-pyrazoles) Contaning Ethynyl Fragments. Russ. J. Org. Chem. 2006, 42, 1368–1373. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, Z.; Lan, D.; Jia, Q.; Liu, N.; Zhang, J.; Kou, K. Recent Advances in Synthesis and Properties of Nitrated-Pyrazoles Based Energetic Compounds. Molecules 2020, 25, 3475. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Tatsui, Y.; Yoneyama, H.; Harusawa, S. C4-Alkylamination of C4-Halo-1H-1-tritylpyrazoles Using Pd(dba)2 or CuI. Molecules 2020, 25, 4634. [Google Scholar] [CrossRef]
- Liu, Q.; Song, Y.; Ma, Y.; Zhou, Y.; Cong, H.; Wang, C.; Wu, J.; Hu, G.; O’Keeffe, M.; Deng, H. Mesoporous Cages in Chemically Robust MOFs Created by a Large Number of Vertices with Reduced Connectivity. J. Am. Chem. Soc. 2019, 141, 488–496. [Google Scholar] [CrossRef]
- Jia, Y.-Y.; Ren, G.-J.; Li, A.-L.; Zhang, L.-Z.; Feng, R.; Zhang, Y.-H.; Bu, X.-H. Temperature-Related Synthesis of Two Anionic Metal–Organic Frameworks with Distinct Performance in Organic Dye Adsorption. Cryst. Growth Des. 2016, 16, 5593–5597. [Google Scholar] [CrossRef]
- Wang, L.-D.; Tao, F.; Cheng, M.-L.; Liu, Q.; Han, W.; Wu, Y.-J.; Yang, D.-D.; Wang, L.-J. Syntheses, Crystal Structures, and Luminescence of Two Main-Group Metal Complexes Based on 3,4-pyrazoledicarboxylic Acid. J. Coord. Chem. 2012, 65, 923–933. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.-B.; Gong, Y.-N.; Zhong, J.-M.; Wen, H.-L. Syntheses, Crystal Structures and Antibacterial Activities of Six Cobalt(II) Pyrazole Carboxylate Complexes with Helical Character. Polyhedron 2012, 36, 6–14. [Google Scholar] [CrossRef]
- Li, F.-L.; Chen, Q.; Song, H.-B.; Dai, B.; Tang, L.-F. Synthesis of Organotin bis(pyrazol-1-yl)methane-tetracarboxylates and Tris(pyrazol-1-yl)methane-hexacarboxylates. Polyhedron 2014, 83, 102–107. [Google Scholar] [CrossRef]
- Cheng, M.; Wang, Q.; Bao, J.; Wu, Y.; Sun, L.; Yang, B.; Liu, Q. Synthesis and Structural Diversity of d10 Metal Coordination Polymers Constructed from New Semi-Rigid Bis(3-methyl-1H-pyrazole-4-carboxylic acid)alkane Ligands. New J. Chem. 2017, 41, 5151–5160. [Google Scholar] [CrossRef]
- Radi, S.; El-Massaoudi, M.; Benaissa, H.; Adarsh, N.N.; Ferbinteanu, M.; Devlin, E.; Sanakis, Y.; Garcia, Y. Crystal Engineering of a Series of Complexes and Coordination Polymers Based on Pyrazole-carboxylic Acid Ligands. New J. Chem. 2017, 41, 8232–8241. [Google Scholar] [CrossRef]
- Kivi, C.E.; Gelfand, B.S.; Dureckova, H.; Ho, H.T.K.; Ma, C.; Shimizu, G.K.H.; Woo, T.K.; Song, D. 3D Porous Metal-Organic Framework for Selective Adsorption of Methane over Dinitrogen under Ambient Pressure. Chem. Commun. 2018, 54, 14104–14107. [Google Scholar] [CrossRef]
- Bloch, W.M.; Burgun, A.; Coghlan, C.J.; Lee, R.; Coote, M.L.; Doonan, C.J.; Sumby, C.J. Capturing Snapshots of Post-Synthetic Metallation Chemistry in Metal-Organic Frameworks. Nat. Chem. 2014, 6, 906. [Google Scholar] [CrossRef]
- Burgun, A.; Coghlan, C.J.; Huang, D.M.; Chen, W.; Horike, S.; Kitagawa, S.; Alvino, J.F.; Metha, G.F.; Sumby, C.J.; Doonan, C.J. Mapping-Out Catalytic Processes in a Metal-Organic Framework with Single-Crystal X-ray Crystallography. Angew. Chem. Int. Ed. 2017, 56, 8412–8416. [Google Scholar] [CrossRef] [Green Version]
- Chiriac, C.I. The Direct Carboxylation of Pyrazoles. Synthesis 1986, 1986, 753–755. [Google Scholar] [CrossRef]
- Padial, N.M.; Quartapelle Procopio, E.; Montoro, C.; López, E.; Oltra, J.E.; Colombo, V.; Maspero, A.; Masciocchi, N.; Galli, S.; Senkovska, I.; et al. Highly Hydrophobic Isoreticular Porous Metal-Organic Frameworks for the Capture of Harmful Volatile Organic Compounds. Angew. Chem. Int. Ed. 2013, 52, 8290–8294. [Google Scholar] [CrossRef]
- Zhao, B.; Liang, Q.; Ren, H.; Zhang, X.; Wu, Y.; Zhang, K.; Ma, L.-Y.; Zheng, Y.-C.; Liu, H.-M. Discovery of Pyrazole Derivatives as Cellular Active Inhibitors of Histone Lysine Specific Demethylase 5B (KDM5B/JARID1B). Eur. J. Med. Chem. 2020, 192, 112161. [Google Scholar] [CrossRef]
- Batista, D.C.; Silva, D.P.B.; Florentino, I.F.; Cardoso, C.S.; Gonçalves, M.P.; Valadares, M.C.; Lião, L.M.; Sanz, G.; Vaz, B.G.; Costa, E.A.; et al. Anti-Inflammatory Effect of a New Piperazine Derivative: (4-methylpiperazin-1-yl)(1-phenyl-1H-pyrazol-4-yl)methanone. Inflammopharmacology 2018, 26, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Hüttel, R.; Schön, M.E. Über Pyrazolyl-lithium-Verbindungen. Justus Liebigs Ann. Chem. 1959, 625, 55–65. [Google Scholar] [CrossRef]
- Janin, Y.L. Synthetic Accesses to 3/5-pyrazole Carboxylic Acids. Mini Rev. Org. Chem. 2010, 7, 314–323. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Torres, J.; Lavandera, J.L.; Cabildo, P.; Claramunt, R.M.; Elguero, J. Synthesis and Physicochemical Studies on 1,2-bisazolylethanes. J. Heterocycl. Chem. 1988, 25, 771–782. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]
- Skidmore, J.; Heer, J.; Johnson, C.N.; Norton, D.; Redshaw, S.; Sweeting, J.; Hurst, D.; Cridland, A.; Vesey, D.; Wall, I.; et al. Optimization of Sphingosine-1-phosphate-1 Receptor Agonists: Effects of Acidic, Basic, and Zwitterionic Chemotypes on Pharmacokinetic and Pharmacodynamic Profiles. J. Med. Chem. 2014, 57, 10424–10442. [Google Scholar] [CrossRef]
- Potapov, A.S.; Khlebnikov, A.I. Synthesis of Mixed-Ligand Copper(II) Complexes Containing Bis(pyrazol-1-yl)methane Ligands. Polyhedron 2006, 25, 2683–2690. [Google Scholar] [CrossRef]
- Potapov, A.S.; Domina, G.A.; Khlebnikov, A.I.; Ogorodnikov, V.D. Facile Synthesis of Flexible bis(pyrazol-1-yl)alkane and Related Ligands in a Superbasic Medium. Eur. J. Org. Chem. 2007, 5112–5116. [Google Scholar] [CrossRef]
- Zatonskaya, L.V.; Schepetkin, I.A.; Petrenko, T.V.; Ogorodnikov, V.D.; Khlebnikov, A.I.; Potapov, A.S. Synthesis and Cytotoxicity of Bis(pyrazol-1-yl)-Alkane Derivatives with Polymethylene Linkers and Related Mono- and Dipyrazolium Salts. Chem. Heterocycl. Compd. 2016, 52, 388–401. [Google Scholar] [CrossRef]





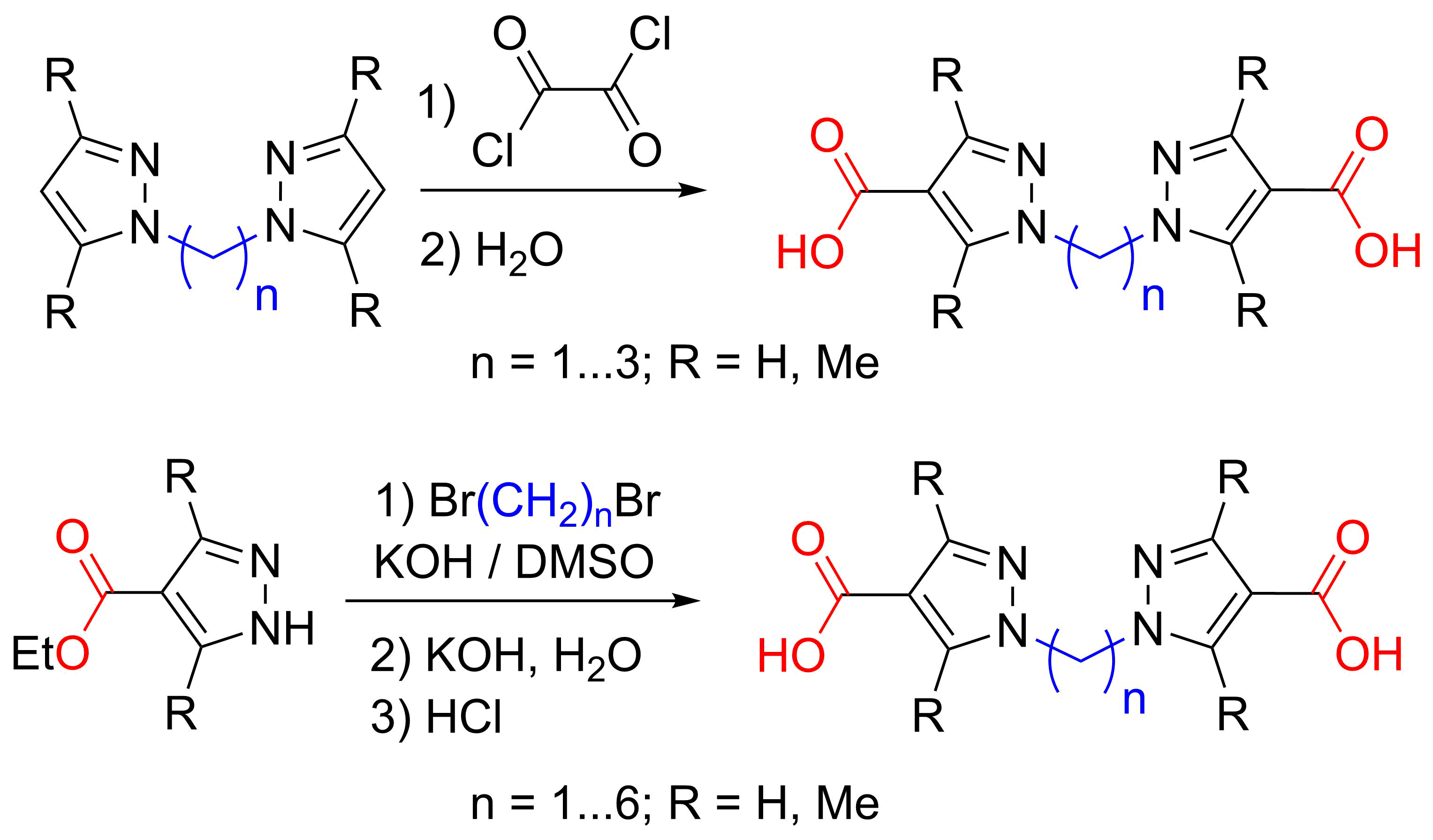{kind=link}
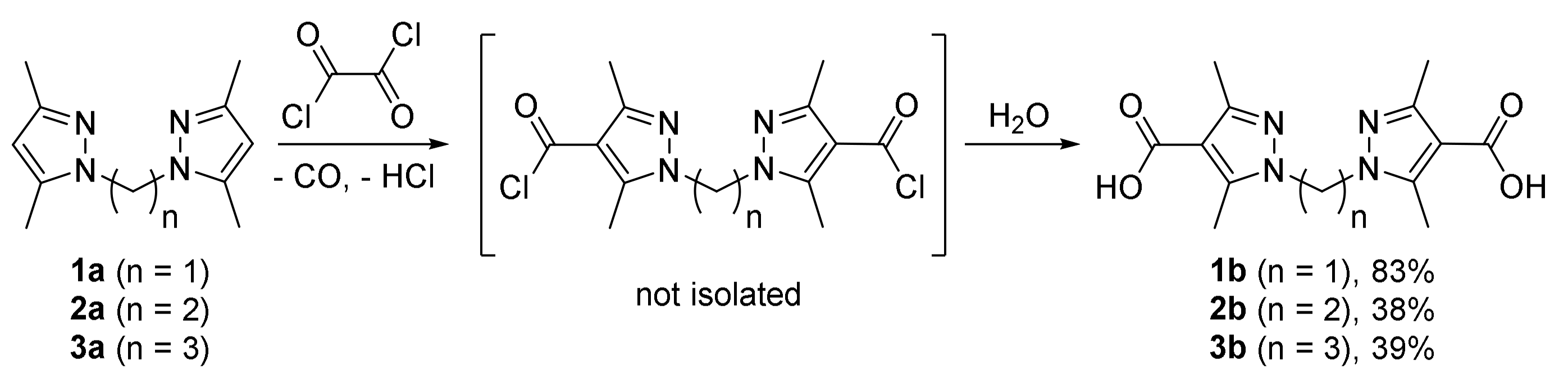{kind=link}
{kind=link}
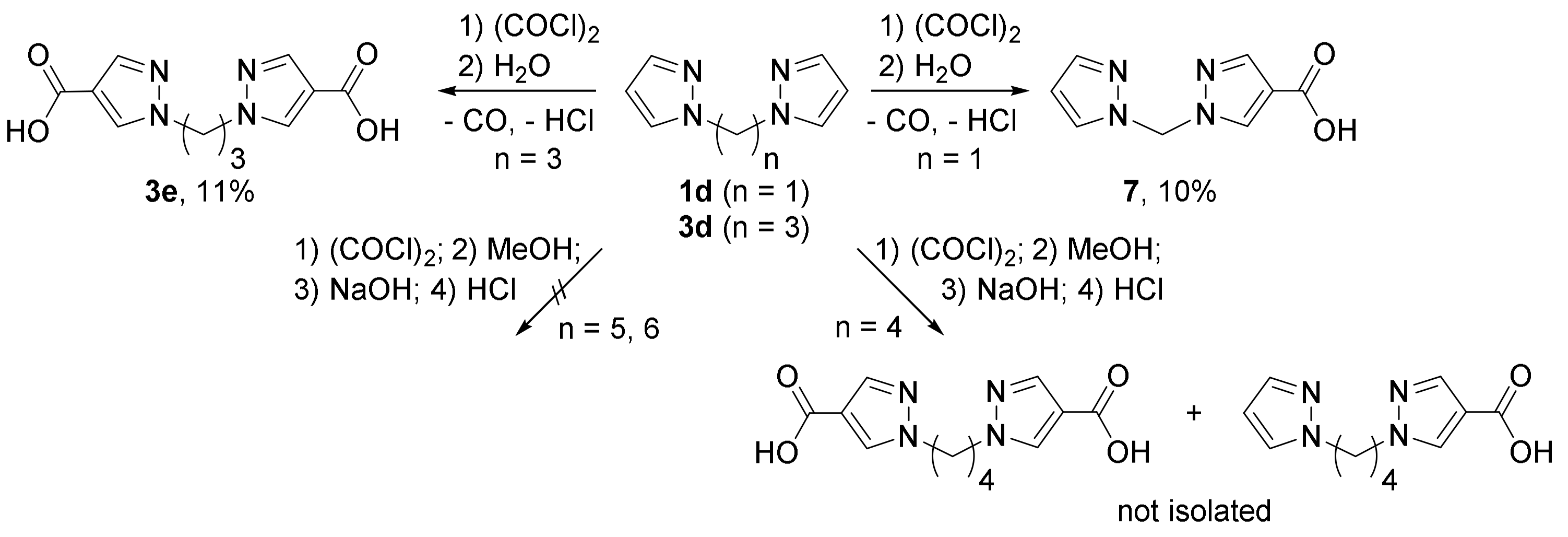{kind=link}
{kind=link}
| Structure | qC | qH | qCH1 | ΔqC 2 | ΔqCH 2 |
|---|---|---|---|---|---|
 | −0.09691 | +0.07896 | −0.01795 | 0.00493 | 0.01012 |
 | −0.09199 | +0.08416 | −0.00783 | ||
 | −0.10241 | +0.07636 | −0.02606 | 0.00349 | 0.00682 |
 | −0.09901 | +0.07978 | −0.01924 | ||
 | −0.07525 | +0.09219 | +0.01694 | 0.00609 | 0.01195 |
 | −0.06916 | +0.09805 | +0.02889 | ||
 | −0.08297 | +0.08930 | +0.00633 | 0.01016 | 0.00904 |
 | −0.07775 | +0.09313 | +0.01538 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlutskiy, N.P.; Potapov, A.S. Approaches to the Synthesis of Dicarboxylic Derivatives of Bis(pyrazol-1-yl)alkanes. Molecules 2021, 26, 413. https://doi.org/10.3390/molecules26020413
Burlutskiy NP, Potapov AS. Approaches to the Synthesis of Dicarboxylic Derivatives of Bis(pyrazol-1-yl)alkanes. Molecules. 2021; 26(2):413. https://doi.org/10.3390/molecules26020413
Chicago/Turabian StyleBurlutskiy, Nikita P., and Andrei S. Potapov. 2021. "Approaches to the Synthesis of Dicarboxylic Derivatives of Bis(pyrazol-1-yl)alkanes" Molecules 26, no. 2: 413. https://doi.org/10.3390/molecules26020413
APA StyleBurlutskiy, N. P., & Potapov, A. S. (2021). Approaches to the Synthesis of Dicarboxylic Derivatives of Bis(pyrazol-1-yl)alkanes. Molecules, 26(2), 413. https://doi.org/10.3390/molecules26020413