
Unravelling the Allosteric Targeting of PHGDH at the ACT-Binding Domain with a Photoactivatable Diazirine Probe and Mass Spectrometry Experiments †

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
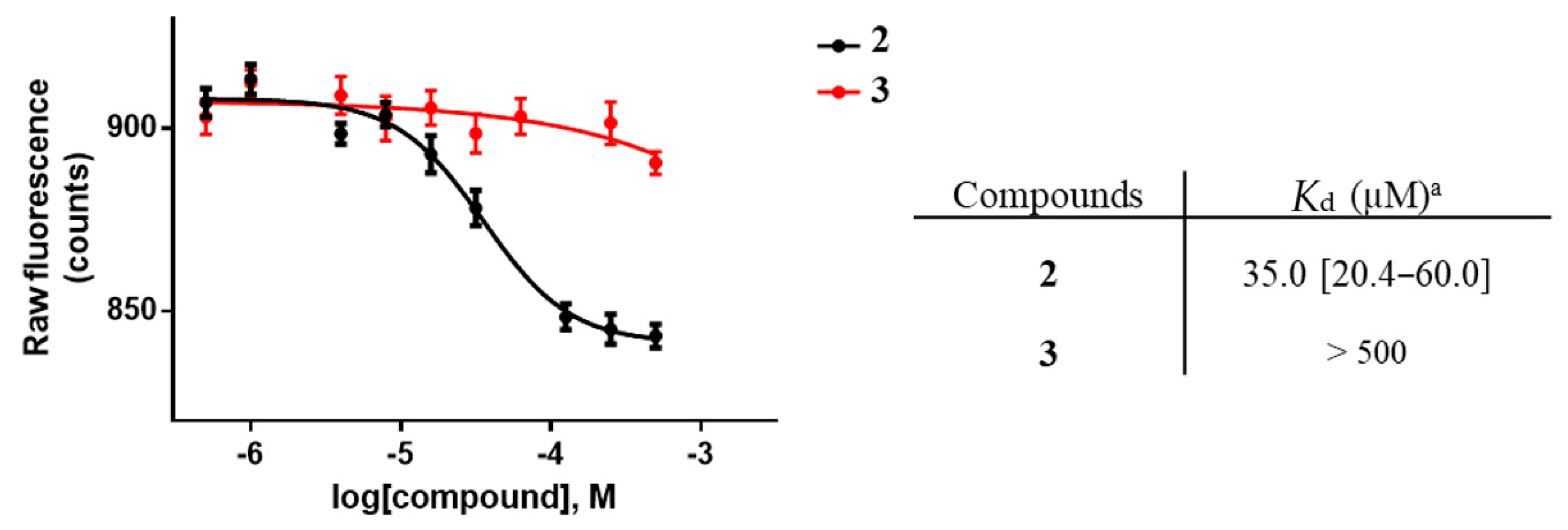
2.1. Biophysical Characterization of the Lead Compound
2.2. Design and Synthesis of the Photoactivatable Probe 11
2.3. Evaluation of the Photoactivable Probe 11
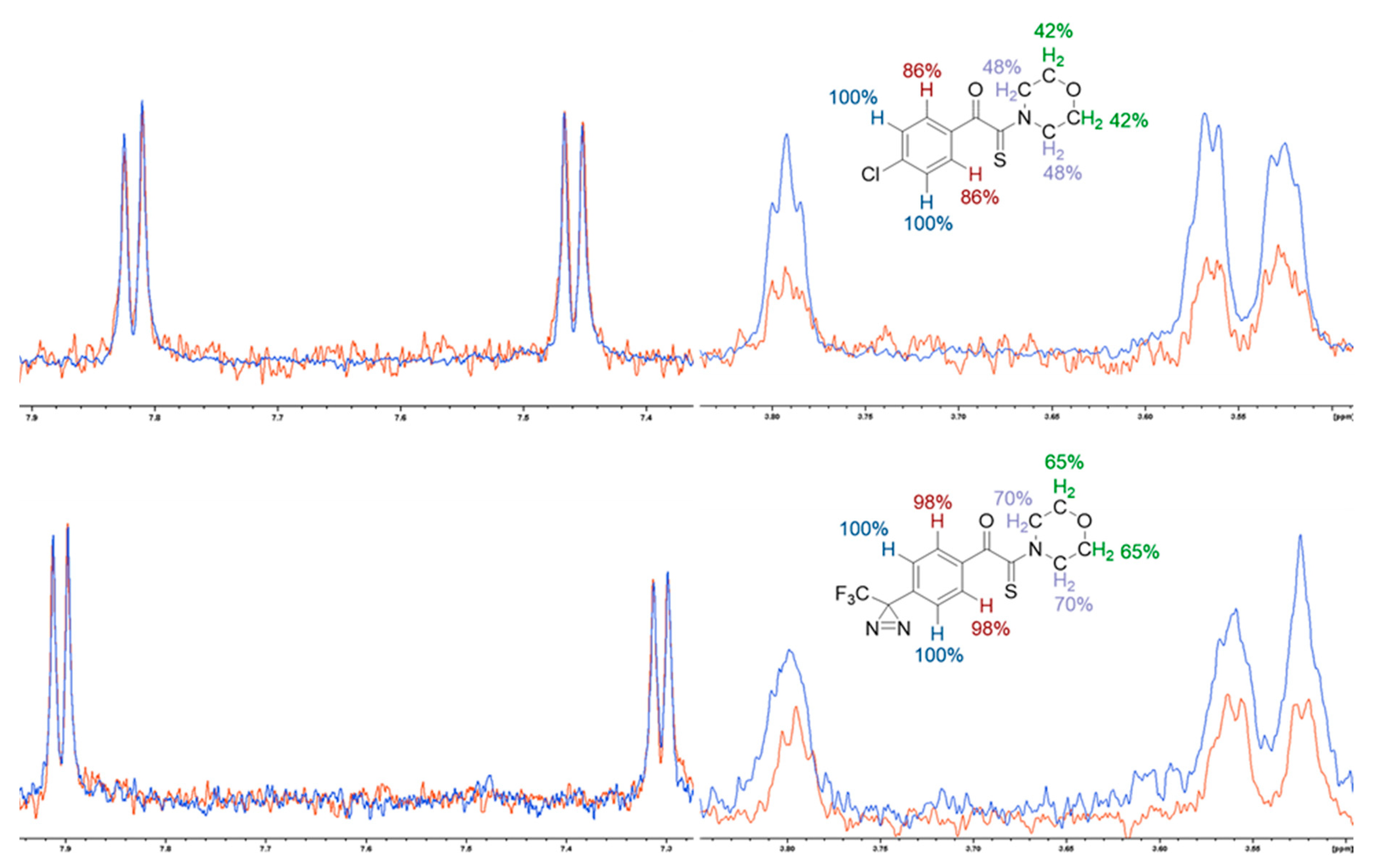
2.4. Epitope Mapping Analysis
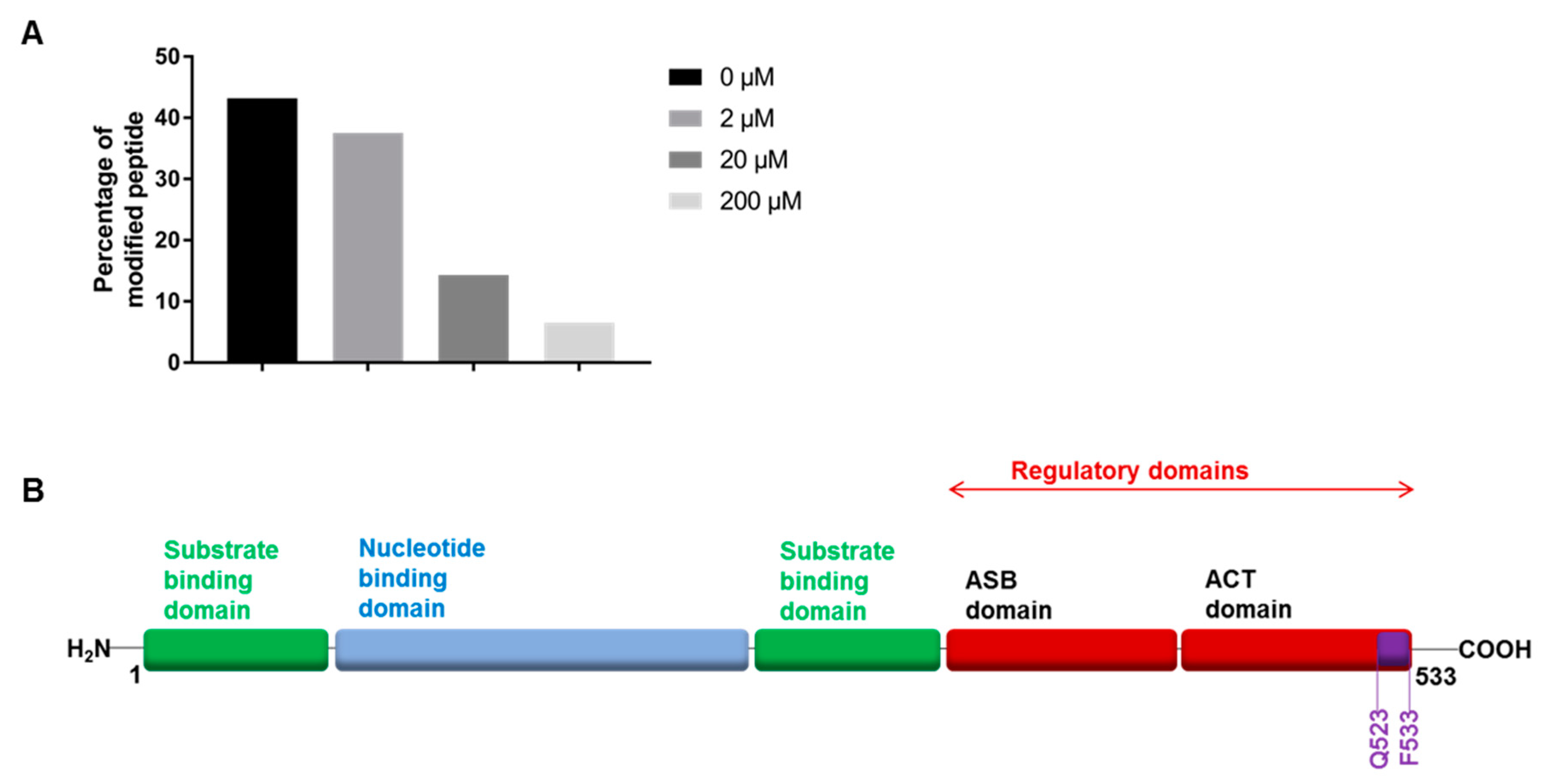
2.5. Photoactivation and Mass Spectrometry Analysis
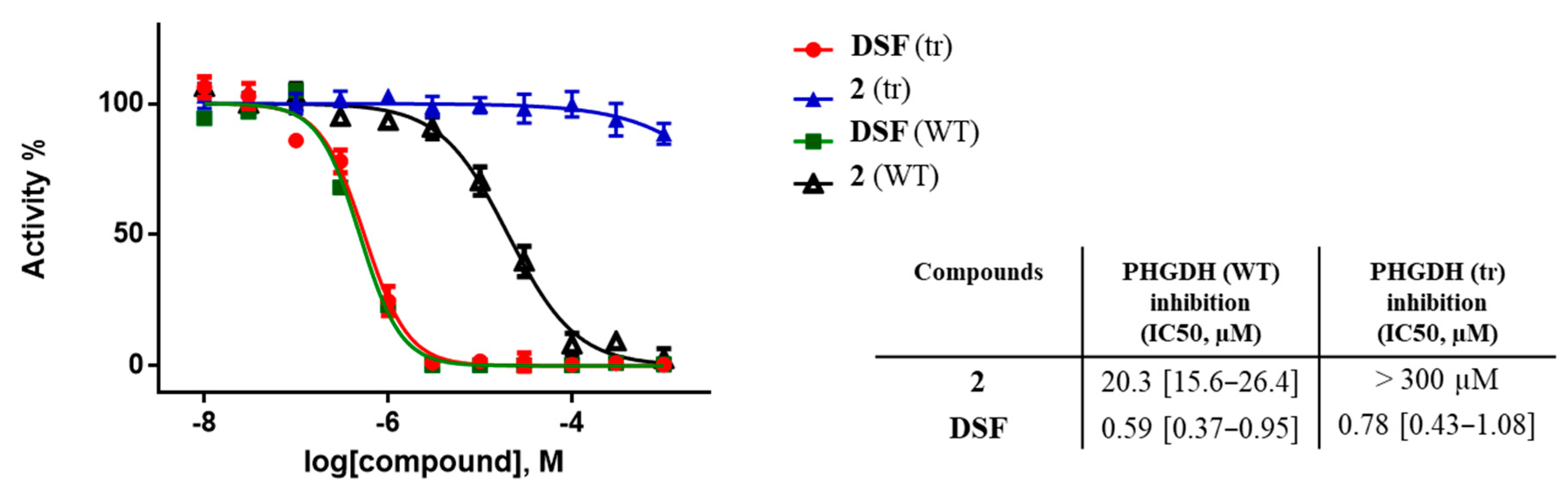
2.6. Protein Truncation Experiments
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Snell, K.; Weber, G. Enzymic imbalance in serine metabolism in rat hepatomas. Biochem. J. 1986, 233, 617–620. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Pollari, S.; Käkönen, S.-M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Guo, S.; Li, Q.; Yang, L.; Xia, Z.; Zhang, L.; Huang, Z.; Zhang, N. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J. Neurooncol. 2013, 111, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Feng, C.; Lu, Y.; Lin, Y.; Dong, C. PHGDH is an independent prognosis marker and contributes cell proliferation, migration and invasion in human pancreatic cancer. Gene 2018, 642, 43–50. [Google Scholar] [CrossRef]
- Gromova, I.; Gromov, P.; Honma, N.; Kumar, S.; Rimm, D.; Talman, M.-L.M.; Wielenga, V.T.; Moreira, J.M.A. High level PHGDH expression in breast is predominantly associated with keratin 5-positive cell lineage independently of malignancy. Mol. Oncol. 2015, 9, 1636–1654. [Google Scholar] [CrossRef]
- Ravez, S.; Spillier, Q.; Marteau, R.; Feron, O.; Frédérick, R. Challenges and Opportunities in the Development of Serine Synthetic Pathway Inhibitors for Cancer Therapy. J. Med. Chem. 2017, 60, 1227–1237. [Google Scholar] [CrossRef]
- Mullarky, E.; Lucki, N.C.; Beheshti Zavareh, R.; Anglin, J.L.; Gomes, A.P.; Nicolay, B.N.; Wong, J.C.Y.; Christen, S.; Takahashi, H.; Singh, P.K.; et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.Y.M.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liberti, M.V.; Liu, P.; Deng, X.; Liu, Y.; Locasale, J.W.; Lai, L. Rational Design of Selective Allosteric Inhibitors of PHGDH and Serine Synthesis with Anti-tumor Activity. Cell Chem. Biol. 2016, 24, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ravez, S.; Corbet, C.; Spillier, Q.; Dutu, A.; Robin, A.D.; Mullarky, E.; Cantley, L.C.; Feron, O.; Frédérick, R. α-Ketothioamide Derivatives: A Promising Tool to Interrogate Phosphoglycerate Dehydrogenase (PHGDH). J. Med. Chem. 2017, 60, 1591–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillier, Q.; Vertommen, D.; Ravez, S.; Marteau, R.; Thémans, Q.; Corbet, C.; Feron, O.; Wouters, J.; Frédérick, R. Anti-alcohol abuse drug disulfiram inhibits human PHGDH via disruption of its active tetrameric form through a specific cysteine oxidation. Sci. Rep. 2019, 9, 4737. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Gu, X.; Zheng, M.; Zhang, Y.; Chen, L.; Li, H. Azacoccone E inhibits cancer cell growth by targeting 3-phosphoglycerate dehydrogenase. Bioorg. Chem. 2019, 87, 16–22. [Google Scholar] [CrossRef]
- Zheng, M.; Guo, J.; Xu, J.; Yang, K.; Tang, R.; Gu, X.; Li, H.; Chen, L. Ixocarpalactone A from dietary tomatillo inhibits pancreatic cancer growth by targeting PHGDH. Food Funct. 2019, 10, 3386–3395. [Google Scholar] [CrossRef]
- Spillier, Q.; Ravez, S.; Unterlass, J.; Corbet, C.; Degavre, C.; Feron, O.; Frédérick, R. Structure–Activity Relationships (SARs) of α-Ketothioamides as Inhibitors of Phosphoglycerate Dehydrogenase (PHGDH). Pharmaceuticals 2020, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.R.; Robertson, A.A.B. Fishing for Drug Targets: A Focus on Diazirine Photoaffinity Probe Synthesis. J. Med. Chem. 2018, 61, 6945–6963. [Google Scholar] [CrossRef]
- Dey, S.; Hu, Z.; Xiao, L.X.; Sacchettini, J.C.; Grant, G.A. D-3-phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J. Biol. Chem. 2005, 280, 14884–14891. [Google Scholar] [CrossRef] [Green Version]
- Thabault, L.; Brisson, L.; Brustenga, C.; Martinez Gache, S.A.; Prévost, J.R.C.; Kozlova, A.; Spillier, Q.; Liberelle, M.; Benyahia, Z.; Messens, J.; et al. Interrogating the lactate dehydrogenase tetramerization site using (stapled) peptides. J. Med. Chem. 2020, 63, 4628–4643. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spillier, Q.; Ravez, S.; Dochain, S.; Vertommen, D.; Thabault, L.; Feron, O.; Frédérick, R. Unravelling the Allosteric Targeting of PHGDH at the ACT-Binding Domain with a Photoactivatable Diazirine Probe and Mass Spectrometry Experiments. Molecules 2021, 26, 477. https://doi.org/10.3390/molecules26020477
Spillier Q, Ravez S, Dochain S, Vertommen D, Thabault L, Feron O, Frédérick R. Unravelling the Allosteric Targeting of PHGDH at the ACT-Binding Domain with a Photoactivatable Diazirine Probe and Mass Spectrometry Experiments. Molecules. 2021; 26(2):477. https://doi.org/10.3390/molecules26020477
Chicago/Turabian StyleSpillier, Quentin, Séverine Ravez, Simon Dochain, Didier Vertommen, Léopold Thabault, Olivier Feron, and Raphaël Frédérick. 2021. "Unravelling the Allosteric Targeting of PHGDH at the ACT-Binding Domain with a Photoactivatable Diazirine Probe and Mass Spectrometry Experiments" Molecules 26, no. 2: 477. https://doi.org/10.3390/molecules26020477
APA StyleSpillier, Q., Ravez, S., Dochain, S., Vertommen, D., Thabault, L., Feron, O., & Frédérick, R. (2021). Unravelling the Allosteric Targeting of PHGDH at the ACT-Binding Domain with a Photoactivatable Diazirine Probe and Mass Spectrometry Experiments. Molecules, 26(2), 477. https://doi.org/10.3390/molecules26020477