
Two Sides of Quantum-Based Modeling of Enzyme-Catalyzed Reactions: Mechanistic and Electronic Structure Aspects of the Hydrolysis by Glutamate Carboxypeptidase


Abstract
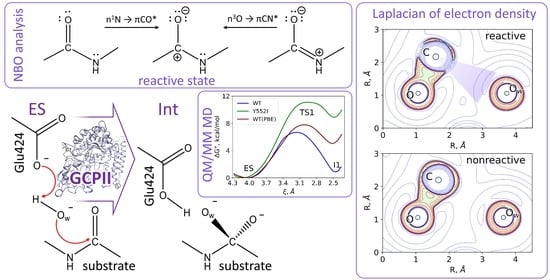
:
1. Introduction
2. Models and Methods
3. Results
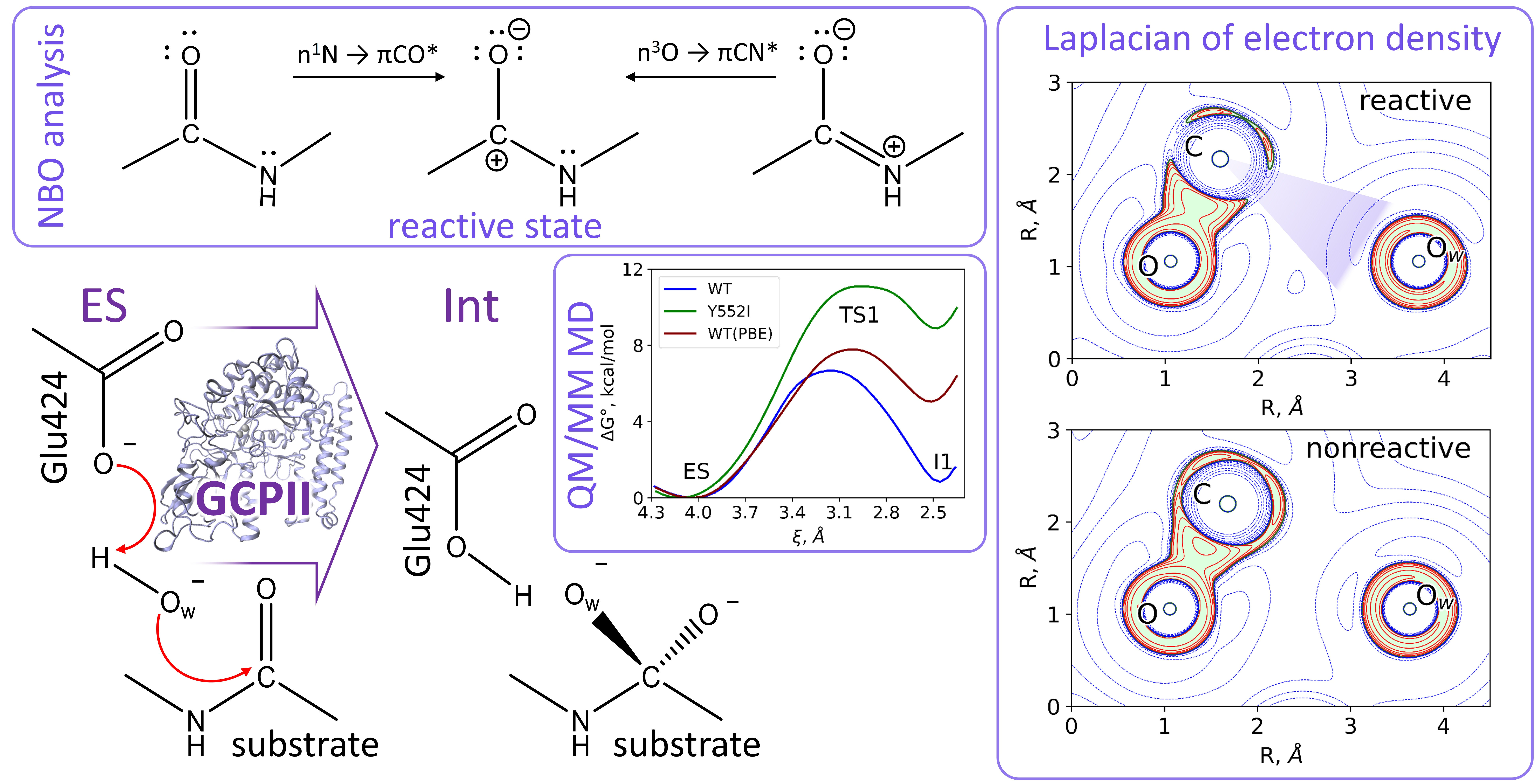
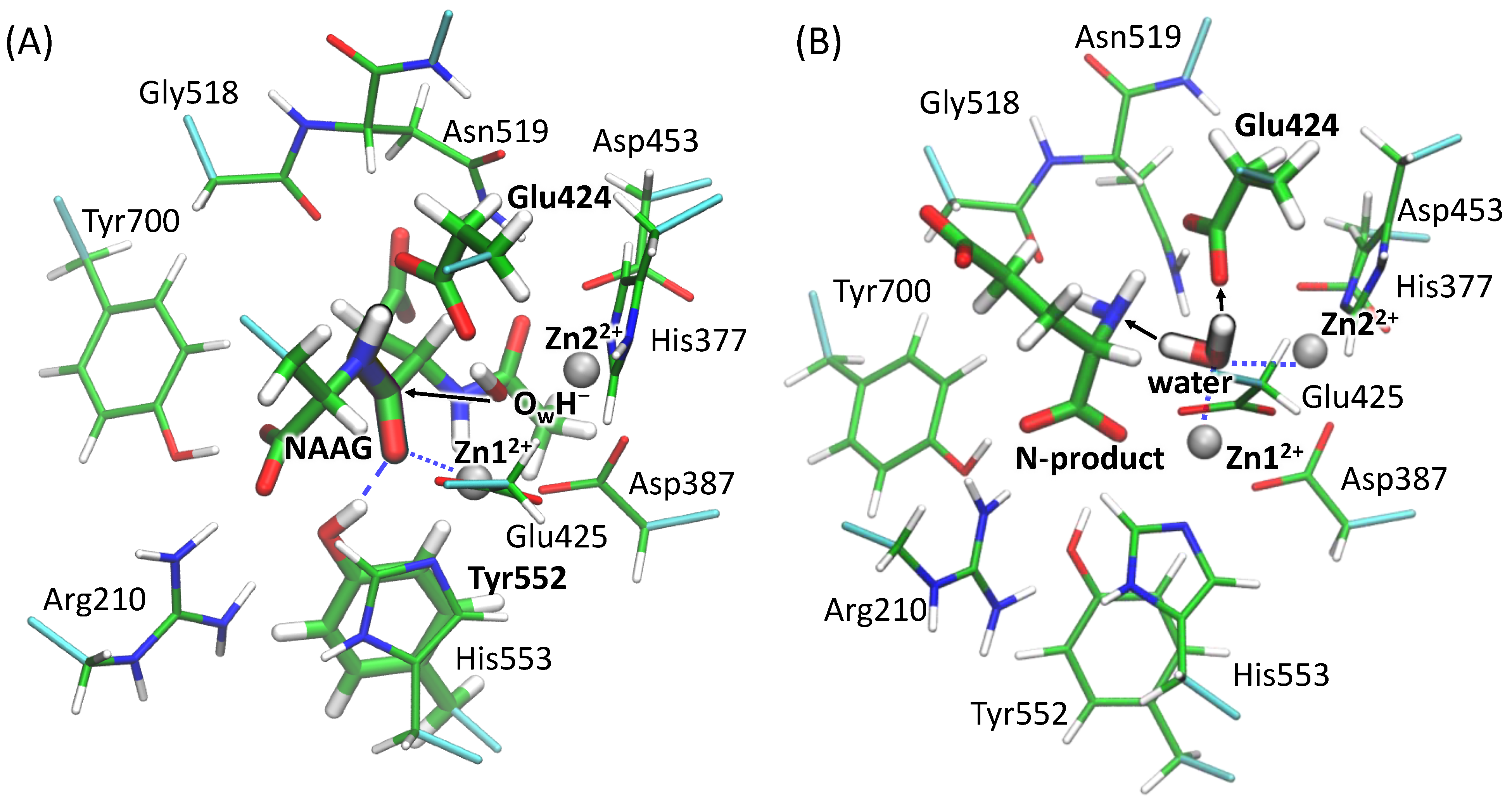
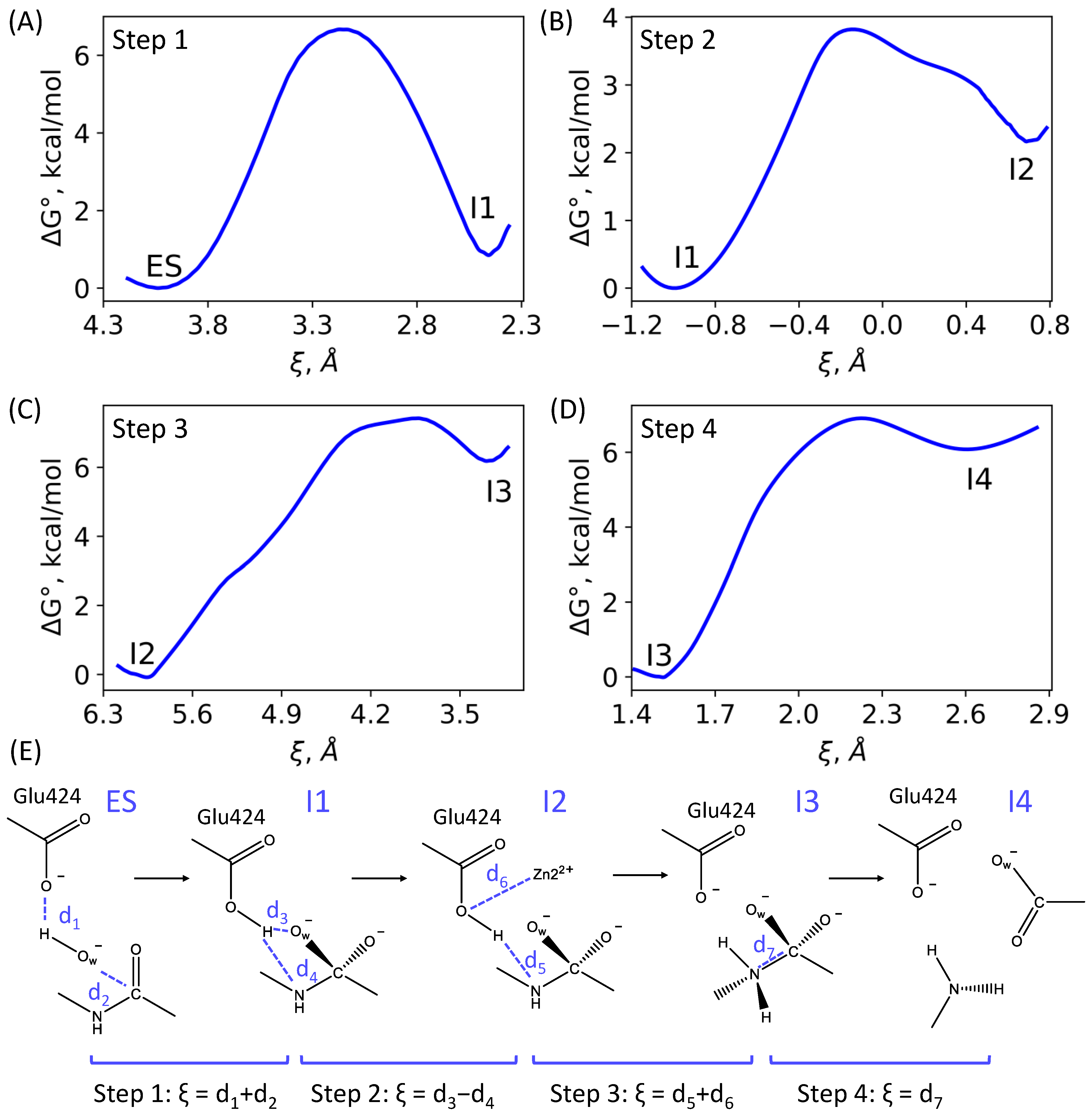
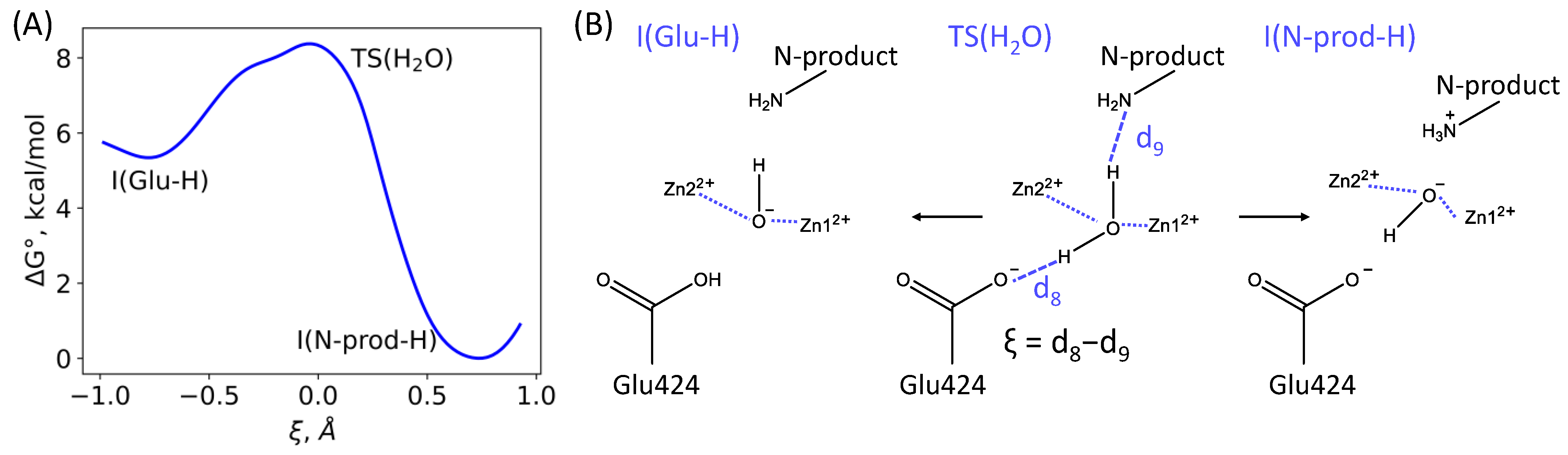
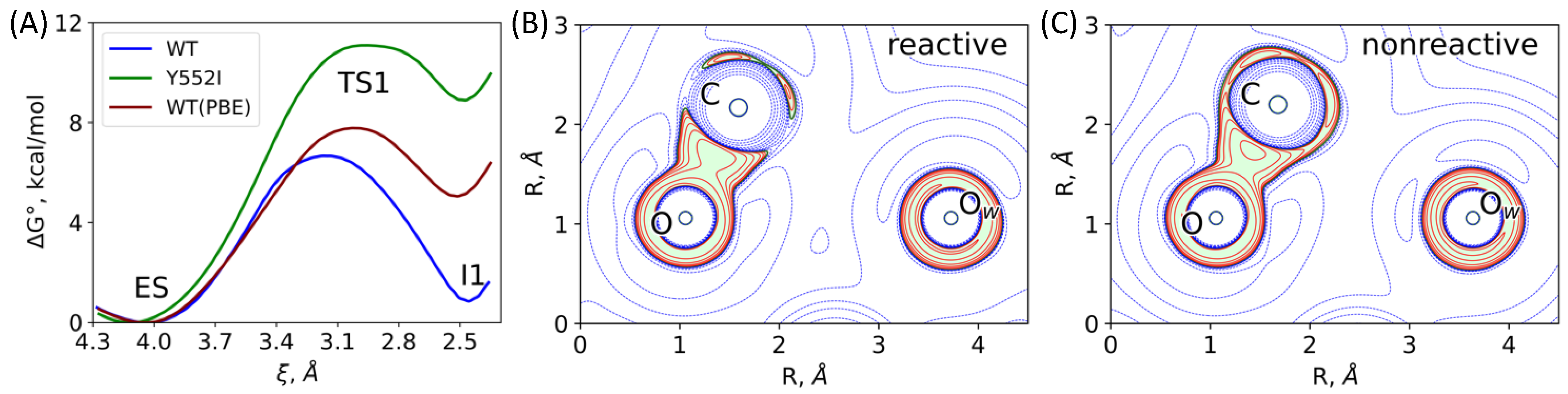
3.1. Mechanism of the NAAG Hydrolysis by CGPII
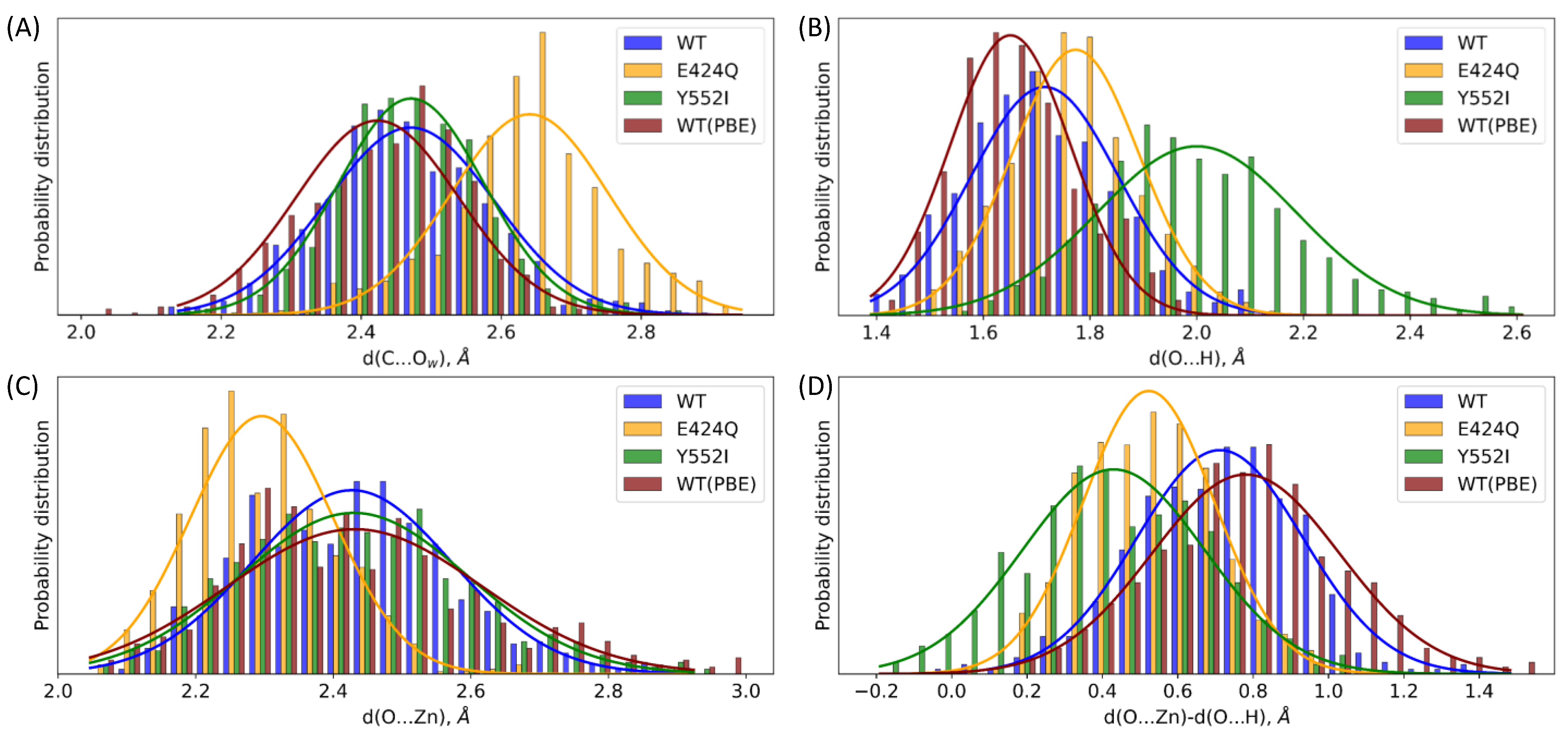
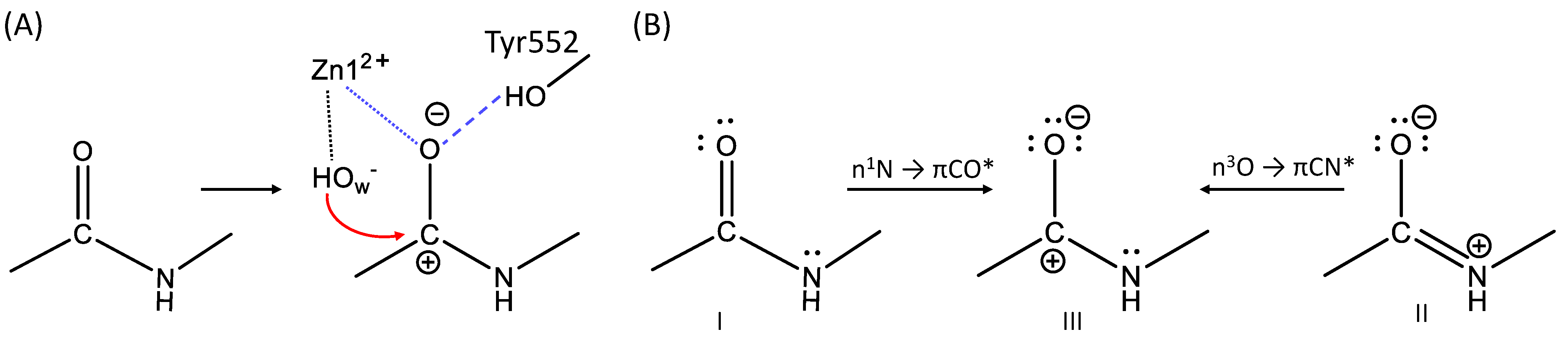
3.2. The First Reaction Step: Reactive and Non-Reactive ES Complexes
3.3. The First Reaction Step: The NBO Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Weinhold, F. The Path to Natural Bond Orbitals. Isr. J. Chem. 2021. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Pauling’s Conceptions of Hybridization and Resonance in Modern Quantum Chemistry. Molecules 2021, 26, 4110. [Google Scholar] [CrossRef] [PubMed]
- Kamerlin, S.C.L.; Warshel, A. The empirical valence bond model: Theory and applications. WIREs Comput. Mol. Sci. 2011, 1, 30–45. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Warshel, A. The EVB as a quantitative tool for formulating simulations and analyzing biological and chemical reactions. Faraday Discuss. 2010, 145, 71–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åqvist, J.; Warshel, A. Simulation of enzyme reactions using valence bond force fields and other hybrid quantum/classical approaches. Chem. Rev. 1993, 93, 2523–2544. [Google Scholar] [CrossRef]
- Lever, G.; Cole, D.J.; Lonsdale, R.; Ranaghan, K.E.; Wales, D.J.; Mulholland, A.J.; Skylaris, C.-K.; Payne, M.C. Large-Scale Density Functional Theory Transition State Searching in Enzymes. J. Phys. Chem. Lett. 2014, 5, 3614–3619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prejanò, M.; Marino, T.; Russo, N. QM Cluster or QM/MM in Computational Enzymology: The Test Case of LigW-Decarboxylase. Front. Chem. 2018, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Prejanò, M.; Alberto, M.E.; Russo, N.; Toscano, M.; Marino, T. The Effects of the Metal Ion Substitution into the Active Site of Metalloenzymes: A Theoretical Insight on Some Selected Cases. Catalysts 2020, 10, 1038. [Google Scholar] [CrossRef]
- Stuyver, T.; De Proft, F.; Geerlings, P.; Shaik, S. How Do Local Reactivity Descriptors Shape the Potential Energy Surface Associated with Chemical Reactions? The Valence Bond Delocalization Perspective. J. Am. Chem. Soc. 2020, 142, 10102–10113. [Google Scholar] [CrossRef]
- Su, S.L.; Huang, I.P.; Fair, W.R.; Powell, C.T.; Heston, W.D. Alternatively spliced variants of prostate-specific membrane antigen RNA: Ratio of expression as a potential measurement of progression. Cancer Res. 1995, 55, 1441–1443. [Google Scholar] [PubMed]
- Robinson, M.B.; Blakely, R.D.; Couto, R.; Coyle, J.T. Hydrolysis of the brain dipeptide N-acetyl-L-aspartyl-L-glutamate. Identification and characterization of a novel N-acetylated alpha-linked acidic dipeptidase activity from rat brain. J. Biol. Chem. 1987, 262, 14498–14506. [Google Scholar] [PubMed]
- Mesters, J.R.; Barinka, C.; Li, W.; Tsukamoto, T.; Majer, P.; Slusher, B.S.; Konvalinka, J.; Hilgenfeld, R. Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 2006, 25, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Barinka, C.; Hlouchova, K.; Rovenska, M.; Majer, P.; Dauter, M.; Hin, N.; Ko, Y.-S.; Tsukamoto, T.; Slusher, B.S.; Konvalinka, J.; et al. Structural basis of interactions between human glutamate carboxypeptidase II and its substrate analogs. J. Mol. Biol. 2008, 376, 1438–1450. [Google Scholar] [CrossRef] [Green Version]
- Klusák, V.; Bařinka, C.; Plechanovová, A.; Mlčochová, P.; Konvalinka, J.; Rulíšek, L.; Lubkowski, J. Reaction Mechanism of Glutamate Carboxypeptidase II Revealed by Mutagenesis, X-ray Crystallography, and Computational Methods. Biochemistry 2009, 48, 4126–4138. [Google Scholar] [CrossRef]
- Navrátil, M.; Tykvart, J.; Schimer, J.; Pachl, P.; Navrátil, V.; Rokob, T.A.; Hlouchová, K.; Rulíšek, L.; Konvalinka, J. Comparison of human glutamate carboxypeptidases II and III reveals their divergent substrate specificities. FEBS J. 2016, 283, 2528–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speno, H.S.; Luthi-Carter, R.; Macias, W.L.; Valentine, S.L.; Joshi, A.R.; Coyle, J.T. Site-directed mutagenesis of predicted active site residues in glutamate carboxypeptidase II. Mol. Pharmacol. 1999, 55, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlcochová, P.; Plechanovová, A.; Barinka, C.; Mahadevan, D.; Saldanha, J.W.; Rulísek, L.; Konvalinka, J. Mapping of the active site of glutamate carboxypeptidase II by site-directed mutagenesis. FEBS J. 2007, 274, 4731–4741. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.C.R.; Bernardi, R.C.; Rudack, T.; Scheurer, M.; Riplinger, C.; Phillips, J.C.; Maia, J.D.C.; Rocha, G.B.; Ribeiro, J.V.; Stone, J.E.; et al. NAMD goes quantum: An integrative suite for QM/MM simulations. Nat. Methods 2018, 15, 351. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Seritan, S.; Bannwarth, C.; Fales, B.S.; Hohenstein, E.G.; Isborn, C.M.; Kokkila-Schumacher, S.I.L.; Li, X.; Liu, F.; Luehr, N.; Snyder, J.W.; et al. TeraChem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics. WIREs Comput. Mol. Sci. 2020. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Kästner, J.; Thiel, W. Bridging the gap between thermodynamic integration and umbrella sampling provides a novel analysis method: “Umbrella integration”. J. Chem. Phys. 2005, 123, 144104. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Tsirelson, V.G.; Nemukhin, A.V. Dynamical properties of enzyme–substrate complexes disclose substrate specificity of the SARS-CoV-2 main protease as characterized by the electron density descriptors. Phys. Chem. Chem. Phys. 2020, 22, 19069–19079. [Google Scholar] [CrossRef]
- Krivitskaya, A.V.; Khrenova, M.G. Boronic Acids as Prospective Inhibitors of Metallo-β-Lactamases: Efficient Chemical Reaction in the Enzymatic Active Site Revealed by Molecular Modeling. Molecules 2021, 26, 2026. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Kulakova, A.M.; Nemukhin, A.V. Light-Induced Change of Arginine Conformation Modulates the Rate of Adenosine Triphosphate to Cyclic Adenosine Monophosphate Conversion in the Optogenetic System Containing Photoactivated Adenylyl Cyclase. J. Chem. Inf. Model. 2021, 61, 1215–1225. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Bulavko, E.S.; Mulashkin, F.D.; Nemukhin, A.V. Mechanism of Guanosine Triphosphate Hydrolysis by the Visual Proteins Arl3-RP2: Free Energy Reaction Profiles Computed with Ab Initio Type QM/MM Potentials. Molecules 2021, 26, 3998. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Grigorenko, B.L.; Nemukhin, A.V. Molecular Modeling Reveals the Mechanism of Ran-RanGAP-Catalyzed Guanosine Triphosphate Hydrolysis without an Arginine Finger. ACS Catal. 2021, 11, 8985–8998. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Carroll, M.T.; Cheeseman, J.R.; Osman, R.; Weinstein, H. Nucleophilic addition to activated double bonds: Predictions of reactivity from the Laplacian of the charge density. J. Phys. Chem. 1989, 93, 5120–5123. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Krivitskaya, A.V.; Tsirelson, V.G. The QM/MM-QTAIM approach reveals the nature of the different reactivity of cephalosporins in the active site of L1 metallo-β-lactamase. New J. Chem. 2019, 43, 7329–7338. [Google Scholar] [CrossRef]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell, A.D. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Grossfield, A. WHAM: The Weighted Histogram Analysis Method. Version 2.0.9. Available online: http://membrane.urmc.rochester.edu/content/wham (accessed on 12 October 2021).
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Khrenova, M.G.; Kulakova, A.M.; Nemukhin, A.V. Proof of concept for poor inhibitor binding and efficient formation of covalent adducts of KRAS G12C and ARS compounds. Org. Biomol. Chem. 2020, 18, 3069–3081. [Google Scholar] [CrossRef] [PubMed]
- Khrenova, M.G.; Nemukhin, A.V.; Tsirelson, V.G. Discrimination of enzyme–substrate complexes by reactivity using the electron density analysis: Peptide bond hydrolysis by the matrix metalloproteinase-2. Mendeleev Commun. 2020, 30, 583–585. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Nemukhin, A.V. Modeling the Transient Kinetics of the L1 Metallo-β-Lactamase. J. Phys. Chem. B 2018, 122, 1378–1386. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Tsirelson, V.G.; Nemukhin, A.V. Computational Characterization of the Substrate Activation in the Active Site of SARS-CoV-2 Main Protease. Supercomput. Front. Innov. 2020, 7. [Google Scholar] [CrossRef]
- Grigorenko, B.L.; Kots, E.D.; Nemukhin, A.V. Diversity of mechanisms in Ras–GAP catalysis of guanosine triphosphate hydrolysis revealed by molecular modeling. Org. Biomol. Chem. 2019, 17, 4879–4891. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Grigorenko, B.L.; Kolomeisky, A.B.; Nemukhin, A.V. Hydrolysis of guanosine triphosphate (GTP) by the Ras-GAP protein complex: Reaction mechanism and kinetic scheme. J. Phys. Chem. B 2015, 119, 12838–12845. [Google Scholar] [CrossRef] [Green Version]
- Khrenova, M.G.; Nemukhin, A.V.; Domratcheva, T. Photoinduced electron transfer facilitates tautomerization of the conserved signaling glutamine side chain in BLUF protein light sensors. J. Phys. Chem. B 2013, 117, 2369–2377. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geometry Parameter | WT | E424Q | Y552I | WT (PBE) |
|---|---|---|---|---|
| d(C...Ow) | 2.47 ± 0.12 | 2.64 ± 0.11 | 2.47 ± 0.10 | 2.42 ± 0.12 |
| d(O...H) | 1.72 ± 0.14 | 1.77 ± 0.12 | 2.00 ± 0.19 | 1.65 ± 0.11 |
| d(O...Zn) | 2.43 ± 0.15 | 2.30 ± 0.10 | 2.43 ± 0.17 | 2.43 ± 0.18 |
| d(O...Zn) − d(O...H) | 0.71 ± 0.22 | 0.52 ± 0.17 | 0.43 ± 0.24 | 0.78 ± 0.25 |
| d(O...Zn) + d(O...H) | 4.14 ± 0.18 | 4.07 ± 0.14 | 4.43 ± 0.26 | 4.08 ± 0.18 |
| Reactivity | WT | E424Q | Y552I | WT (PBE) |
|---|---|---|---|---|
| Fraction of reactive conformations | 88% | 6% | 0% | 2% |
| Structure Type | WT | E424Q | Y552I | WT (PBE) |
|---|---|---|---|---|
| I | 36% | 20% | 72% | 16% |
| II | 62% | 80% | 28% | 82% |
| III | 2% | 0% | 0% | 2% |
| Structure Type | Interaction Type | WT | E424Q | Y552I | WT (PBE) |
|---|---|---|---|---|---|
| I | nN → πCO* | 63 | 33 | 36 | 34 |
| II | nO → πCN* | 47 | 24 | 26 | 21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivitskaya, A.V.; Khrenova, M.G.; Nemukhin, A.V. Two Sides of Quantum-Based Modeling of Enzyme-Catalyzed Reactions: Mechanistic and Electronic Structure Aspects of the Hydrolysis by Glutamate Carboxypeptidase. Molecules 2021, 26, 6280. https://doi.org/10.3390/molecules26206280
Krivitskaya AV, Khrenova MG, Nemukhin AV. Two Sides of Quantum-Based Modeling of Enzyme-Catalyzed Reactions: Mechanistic and Electronic Structure Aspects of the Hydrolysis by Glutamate Carboxypeptidase. Molecules. 2021; 26(20):6280. https://doi.org/10.3390/molecules26206280
Chicago/Turabian StyleKrivitskaya, Alexandra V., Maria G. Khrenova, and Alexander V. Nemukhin. 2021. "Two Sides of Quantum-Based Modeling of Enzyme-Catalyzed Reactions: Mechanistic and Electronic Structure Aspects of the Hydrolysis by Glutamate Carboxypeptidase" Molecules 26, no. 20: 6280. https://doi.org/10.3390/molecules26206280
APA StyleKrivitskaya, A. V., Khrenova, M. G., & Nemukhin, A. V. (2021). Two Sides of Quantum-Based Modeling of Enzyme-Catalyzed Reactions: Mechanistic and Electronic Structure Aspects of the Hydrolysis by Glutamate Carboxypeptidase. Molecules, 26(20), 6280. https://doi.org/10.3390/molecules26206280