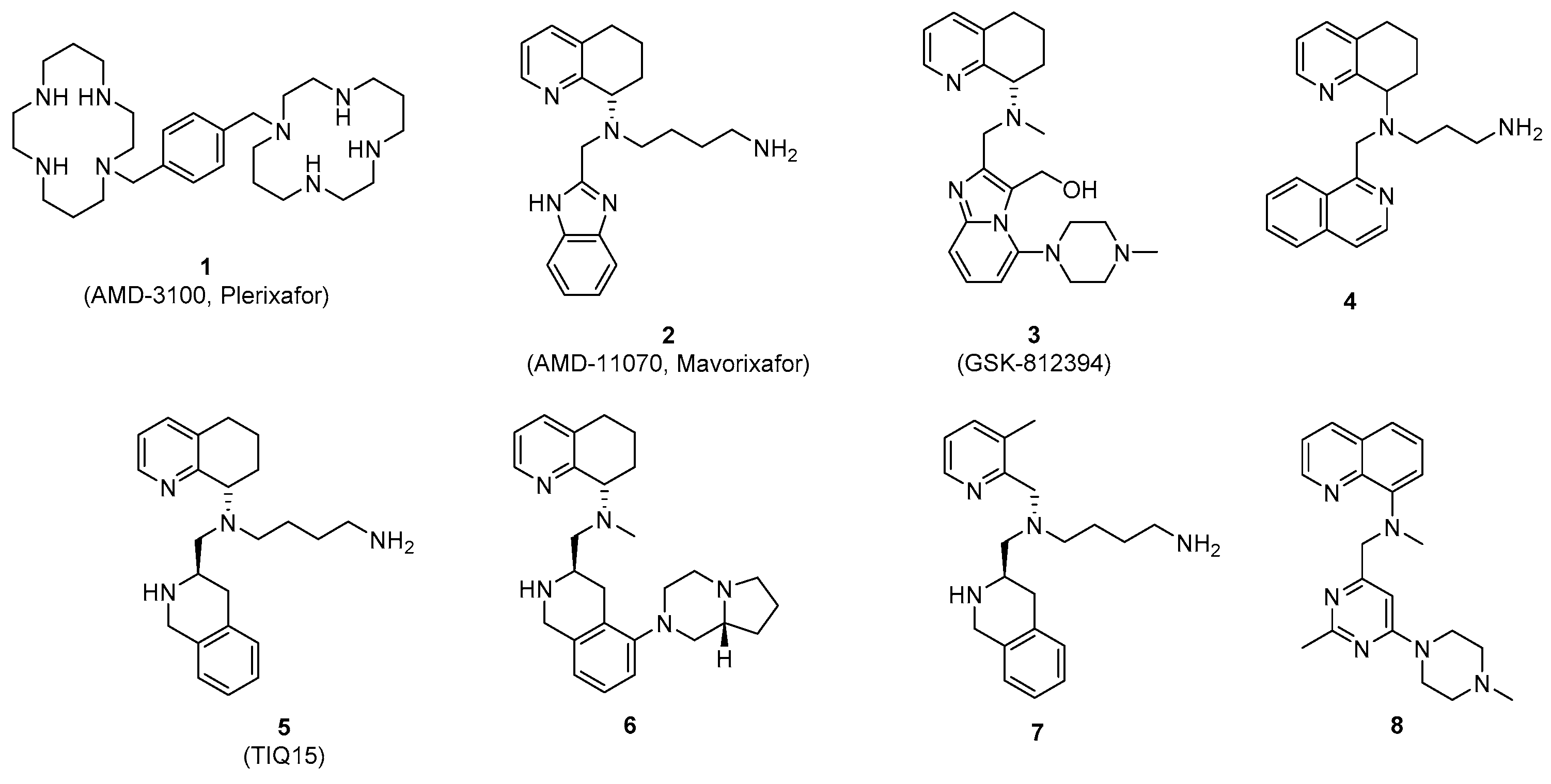
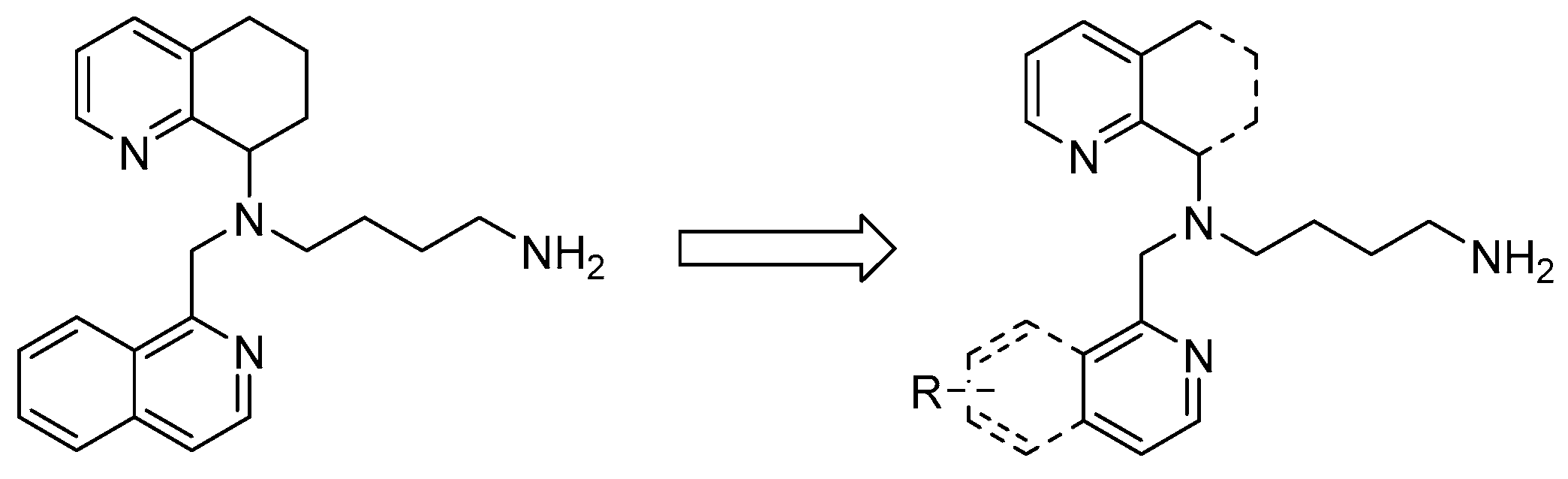
3.2. Chemistry
3.2.1. Synthesis of Dimethyl Acetal-Substituted Triazoles (12a–d)
General procedure
Commercially available substituted acetophenones 9a–d (1 eq., 2.5 mmol) and 2-aminoacetaldehyde dimethyl acetal 10 (1.1 eq., 2.75 mmol, 289 mg) were added to an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar. The mixture was dissolved in dry toluene (2 mL) followed by addition of 4-nitrophenyl azide 11 (1.5 eq., 3.75 mmol, 615 mg) and stirred overnight at 100 °C in an aluminum heating block. The crude residue was purified via silica gel flash chromatography, yielding the title compounds. The following compounds were made according to this general procedure.
1-(2,2-Dimethoxyethyl)-5-(3,4-dimethoxyphenyl)-1H-1,2,3-triazole (12a)
This compound was prepared from 3′,4′-dimethoxyacetophenone 9a (415.45 mg, 2.50 mmol). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (95:5) as mobile phase, yielding the title compound as a yellowish-orange solid (654 mg, 89%); mp 90–92 °C. 1H NMR (400 MHz, CDCl3) δ: 7.67 (s, 1H), 7.10 (d, J = 2.0 Hz, 1H), 7.06 (dd, J = 8.2, 2.0 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 4.93 (t, J = 5.6 Hz, 1H), 4.41 (d, J = 5.7 Hz, 2H), 3.94 (s, 3H), 3.92 (s, 3H), 3.37 (s, 6H). 13C NMR (101 MHz, CDCl3) δ: 150.0, 149.3, 139.0, 132.6, 122.1, 119.2, 112.4, 111.5, 103.7, 56.1, 56.1, 55.4, 49.7. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C14H19N3O4: 294.1448; found: 294.1455.
This compound was prepared from 4′-methoxyacetophenone 9b (375.45 mg, 2.50 mmol). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (95:5) as mobile phase, yielding the title compound as a yellowish-orange viscous oil (488 mg, 74%). 1H NMR (400 MHz, CDCl3) δ: 7.64 (s, 1H), 7.42 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 4.88 (t, J = 5.6 Hz, 1H), 4.39 (d, J = 5.7 Hz, 2H), 3.85 (s, 3H), 3.33 (s, 6H). 13C NMR (101 MHz, CDCl3) δ: 160.4, 138.7, 132.5, 130.5, 118.9, 114.4, 103.3, 55.3, 55.1, 49.4. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C13H17N3O3: 264.1343; found: 264.1346.
This compound was prepared from 4′-fluoroacetophenone 9c (345.35 mg, 2.50 mmol). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (95:5) as the mobile phase, yielding the title compound as a reddish viscous oil (447 mg, 71%). 1H NMR (400 MHz, CDCl3) δ: 7.65 (s, 1H), 7.47 (dd, J = 8.8, 5.2 Hz, 2H), 7.16 (t, J = 8.6 Hz, 2H), 4.86 (t, J = 5.6 Hz, 1H), 4.35 (d, J = 5.6 Hz, 2H), 3.33 (s, 6H). 13C NMR (101 MHz, CDCl3) δ: 164.6, 162.1, 138.0, 132.9, 131.2, 122.9, 116.2, 103.6, 55.5, 49.6. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C12H14FN3O2: 252.1143; found: 252.1139.
This compound was prepared from 4′-bromoacetophenone 9d (497.6 mg, 2.50 mmol). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (95:5) as mobile phase, yielding the title compound as an orange viscous oil (485 mg, 62%). 1H NMR (400 MHz, CDCl3) δ: 7.68 (s, 1H), 7.62 (d, J = 8.5 Hz, 2H), 7.37 (d, J = 8.5 Hz, 2H), 4.87 (t, J = 5.6 Hz, 1H), 4.36 (d, J = 5.6 Hz, 2H), 3.34 (s, 6H). 13C NMR (101 MHz, CDCl3) δ: 138.1, 133, 132.4, 130.9, 125.9, 124.2, 103.7, 55.6, 49.8. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C12H14BrN3O2: 312.0343; found: 312.0338.
3.2.2. Synthesis of 1,2,3-Triazolo[5,1-a]isoquinolines (13a–d)
General procedure
To an open round-bottom flask equipped with a magnetic stirring bar containing the triazole 12a–d (1.2 mmol) was added concentrated H2SO4 (80–95%, 2.5–4 mL), while stirring at 0 °C in an ice bath. After cooling for 30 min, the reaction mixture was stirred overnight at room temperature. The reaction mixture was poured over ice and neutralized by slowly adding a 3 M aqueous NaOH solution. The aqueous phase was subsequently extracted five times with dichloromethane. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The crude residue was purified by flash chromatography, yielding the title compound. The following compounds were made according to this procedure.
8,9-Dimethoxy-[1,2,3]triazolo[5,1-a]isoquinoline (13a)
This compound was prepared from triazole 12a (354 mg, 1.21 mmol). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (80:20) as the mobile phase, yielding the title compound as an off-white solid (274 mg, 99%). mp 221–224 °C. 1H NMR (400 MHz, CDCl3) δ: 8.42 (d, J = 7.3 Hz, 1H), 8.29 (s, 1H), 7.39 (s, 1H), 7.13 (s, 1H), 7.07 (d, J = 7.3 Hz, 1H), 4.05 (s, 3H), 4.02 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 150.7, 150.6, 132.3, 124.5, 123.9, 121.0, 117.3, 115.4, 107.9, 104.5, 56.3, 56.2. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C12H11N3O2: 230.0924; found: 230.0936.
This compound was prepared from triazole 12b (301 mg, 1.14 mmol). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (80:20) as the mobile phase, yielding the title compound as an off-white solid (226 mg, 99%). mp 133–134 °C. 1H NMR (400 MHz, CDCl3) δ: 8.43 (d, J = 7.4 Hz, 1H), 8.26 (s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.20 (dd, J = 8.8, 2.6 Hz, 1H), 7.12 (d, J = 2.5 Hz, 1H), 7.06 (d, J = 7.4 Hz, 1H), 3.92 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 160.0, 132.5, 130.7, 125.5, 124.6, 122.8, 118.3, 116.7, 115.6, 108.8, 55.5. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C11H9N3O: 200.0818; found: 200.0826.
This compound was prepared from triazole 12c (301 mg, 1.2 mmol) and H2SO4 (80%, aq.). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (80:20) as the mobile phase, yielding the title compound as an off-white solid (196 mg, 87%). mp 123–126 °C. 1H NMR (400 MHz, CDCl3) δ: 8.55 (d, J = 7.4 Hz, 1H), 8.38 (s, 1H), 8.15 (dd, J = 8.6, 5.3 Hz, 1H), 7.47 (d, J = 9 Hz, 1H), 7.4 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 7.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 162.71 (d, J = 250.3 Hz), 138.48, 133.55, 130.96 (d, J = 8.5 Hz), 126.62 (d, J = 9.1 Hz), 125.63, 123.90, 117.79 (d, J = 23.9 Hz), 115.42 (d, J = 3.4 Hz), 113.05 (d, J = 22.2 Hz). HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C10H6FN3: 188.0618; found: 188.0617.
This compound was prepared from triazole 12d (375 mg, 1.2 mmol). The crude residue was purified by silica gel flash chromatography using DCM/EtOAc (80:20) as the mobile phase, yielding the title compound as an off-white solid (285 mg, 95%). mp 209–211 °C. 1H NMR (400 MHz, CDCl3) δ: 8.54 (d, J = 7.4 Hz, 1H), 8.42 (s, 1H), 8.02 (d, J = 8.5 Hz, 1H), 7.97 (d, J = 1.9 Hz, 1H), 7.78 (dd, J = 8.5, 1.9 Hz, 1H), 7.3 (d, J = 7.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 131.8, 131.7, 130.3, 129.8, 125.7, 125.3, 123.5, 1229, 121.4, 114.6. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C10H6BrN3: 247.9818; found: 247.9812.
3.2.3. Synthesis of 1-Hydroxymethylisoquinolines (14a–d)
General procedure
A solution of the triazoloisoquinoline 13a–d (1.2 mmol) in a 2.5M H2SO4 solution (8–10 mL) was heated at 120 °C until completion of the reaction (24–36 h). Subsequently, the reaction mixture was poured over ice and neutralized by adding a saturated aqueous NaHCO3 solution. The aqueous phase was extracted three times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated in vacuo to afford the title compound, which was used for the next step without further purification. The following compounds were made according to this procedure.
This compound was prepared from triazoloisoquinoline 13a (275 mg, 1.2 mmol). The title compound was isolated as an off-white solid (242 mg, 92%). mp 131–133 °C. 1H NMR (400 MHz, CDCl3) δ: 8.33 (d, J = 5.7, 1H), 7.46 (d, J = 5.6, 1H), 7.11 (s, 1H), 7.05 (s, 1H), 5.13 (s, 2H), 4.04 (s, 3H), 4.03 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 155.5, 153.7, 151.1, 140.1, 133.5, 119.9, 106.1, 101.9, 62.1, 56.8. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C12H13NO3: 220.0968; found: 220.0974.
This compound was prepared from triazoloisoquinoline 13b (240 mg, 1.2 mmol). The title compound was isolated as an off-white solid (173 mg, 76%); mp 63–65 °C. 1H NMR (400 MHz, CDCl3) δ: 8.30 (d, J = 5.8 Hz, 1H), 7.76 (d, J = 9.2 Hz, 1H), 7.43 (d, J = 5.7 Hz, 1H), 7.19 (s, 1H), 7.04 (s, 1H), 5.11 (s, 2H), 3.88 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 160.7, 156.5, 140.5, 137.9, 124.8, 120.2, 120.1, 119.5, 104.8, 60.9, 55.2. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C11H11NO2: 190.0862; found: 190.0869.
This compound was prepared from triazoloisoquinoline 13c (225 mg, 1.2 mmol). The title compound was isolated as an off-white solid (194 mg, 91%). mp 72–74°C. 1H NMR (400 MHz, CDCl3) δ: 8.45 (d, J = 5.8 Hz, 1H), 7.96 (dd, J = 9.1, 5.3 Hz, 1H), 7.57 (d, J = 5.8 Hz, 1H), 7.48 (dd, J = 9.2, 2.5 Hz, 1H), 7.3 (td, J = 8.5, 2.5 Hz, 1H), 5.22 (s, 2H), 4.93 (s, 1H). 13C NMR (101 MHz, CDCl3) δ: 163.9, 161.4, 156.9, 140.8, 137.1, 125.8, 121.6, 119.4, 117.4, 110.3. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C10H8FNO: 178.0663; found: 178.0659.
This compound was prepared from triazoloisoquinoline 13d (298 mg, 1.2 mmol). The title compound was isolated as an off-white solid (238 mg, 83%). mp 143–145°C. 1H NMR (400 MHz, CDCl3) δ: 8.47 (d, J = 5.4 Hz, 1H), 8.03 (d, J = 1.8 Hz, 1H), 7.79 (d, J = 8.9 Hz, 1H), 7.69 (dd, J = 8.9, 1.9 Hz, 1H), 7.51 (d, J = 5.7 Hz, 1H), 5.20 (s, 2H), 4.89 (s, 1H). 13C NMR (101 MHz, CDCl3) δ: 158.2, 137.5, 131.6, 130, 125.7, 125.4, 123.8, 119.7, 61.8. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C10H8BrNO: 237.9862; found: 237.9851
3.2.4. Synthesis of 1-(Chloromethyl)isoquinolines (15a–d)
General procedure
To a solution of 1-hydroxymethylisoquinoline 14a–d (1.2 mmol) in dry dichloromethane (1 mL) was added an excess of SOCl2. The resulting reaction mixture was stirred for 3 h at room temperature. After completion of the reaction, the reaction mixture was neutralized with an aqueous saturated NaHCO3 solution. The aqueous phase was extracted several times with dichloromethane. The combined organic layers were dried over MgSO4 and concentrated under vacuum to afford the title compound, that was used for the next step without further purification. The following compounds were made according to this procedure.
This compound was prepared from 1-hydroxymethylisoquinoline 14a (263 mg, 1.2 mmol). The title compound was isolated as a red-orange solid; yield: 285 mg (99%); mp 148–150 °C. 1H NMR (400 MHz, CDCl3) δ: 8.34 (d, J = 5.8 Hz, 1H), 7.50 (d, J = 5.5 Hz, 1H), 7.42 (s, 1H), 7.09 (s, 1H), 5.09 (s, 2H), 4.07 (s, 3H), 4.03 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 153.1, 153.0, 150.6, 141.0, 133.8, 122.7, 120.7, 105.4, 103.2, 56.2, 56.2, 45.7. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C12H12ClNO2: 238.0629; found: 238.0631.
This compound was prepared from 1-hydroxymethylisoquinoline 14b (227 mg, 1.2 mmol). The title compound was isolated as a dark red solid (225 mg, 90%). mp 85–87 °C. 1H NMR (400 MHz, CDCl3) δ: 8.40 (d, J = 5.7 Hz, 1H), 8.15 (d, J = 9.2 Hz, 1H), 7.55 (d, J = 5.7 Hz, 1H), 7.29 (dd, J = 9.2, 2.5 Hz, 1H), 7.10 (d, J = 2.5 Hz, 1H), 5.09 (s, 2H), 3.96 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 160.9, 154.7, 141.7, 138.9, 126.8, 121.0, 120.8, 104.8, 55.4, 44.3. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C11H10ClNO: 208.0524; found: 208.0516.
This compound was prepared from 1-hydroxymethylisoquinoline 14c (213 mg, 1.2 mmol). The title compound was isolated as a red solid (224 mg, 95%). mp 65–67 °C. 1H NMR (600 MHz, CDCl3) δ: 8.47 (d, J = 5.7 Hz, 1H), 8.30 (dd, J = 9.1, 5.3 Hz, 1H), 7.62 (d, J = 5.7 Hz, 1H), 7.52–7.39 (m, 2H), 5.12 (s, 2H). 13C NMR (151 MHz, CDCl3) δ: 164, 162.4, 155.9, 142.9, 138.6, 128.5, 123.8, 121.6, 118.5, 110.9, 45. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C10H7ClFN: 196.0324; found: 196.0316.
This compound was prepared from 1-hydroxymethylisoquinoline 14d (286 mg, 1.2 mmol). The title compound was isolated as a reddish solid (301 mg, 98%). mp 108–110 °C. 1H NMR (400 MHz, CDCl3) δ: 8.50 (d, J = 5.7 Hz, 1H), 8.14 (d, J = 9 Hz, 1H), 8.05 (d, J = 1.9 Hz, 1H), 7.75 (dd, J = 9, 1.9 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 5.12 (s, 2H). 13C NMR (101 MHz, CDCl3) δ: 156.1, 143.1, 137.9, 131.5, 129.8, 127, 125.4, 125, 120.8, 44.8. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C10H7BrClN: 255.9524; found: 255.9520.
3.2.5. tert-Butyl (4-aminobutyl) carbamate (17)
To a solution of 1,4-diaminobutane 16 (10 eq., 56.7 mmol, 5 g) in dichloromethane (62.5 mL) was added a solution of Boc2O (1 eq., 5.67 mmol, 1.238 g) in dichloromethane (62.5 mL) through a dropping funnel. The reaction mixture was stirred at room temperature for 16 h. After filtering the suspension, the filtrate was evaporated under vacuum. The oily residue was washed with brine and extracted with EtOAc to remove the excess amount of diamine. The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum, yielding the title compound as a colorless oil (6.9 g, 65%). 1H NMR (400 MHz, CDCl3) δ: 4.77 (s, 1H), 3.07 (t, J = 6.0 Hz, 2H), 2.66 (t, J = 6.6 Hz, 2H), 1.52–1.42 (m, 4H), 1.39 (s, 9H), 1.09 (s, 2H). 13C NMR (75 MHz, CDCl3) δ: 155.88, 78.41, 41.51, 40.09, 30.59, 28.17, 27.19. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C9H20N2O2: 189.1597; found: 189.1597.
3.2.6. tert-Butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl) carbamate (18)
6,7-Dihydroquinolin-8(5H)-one (1.03 eq., 3.40 mmol, 0.500 g) was added to a slurry of NaBH(OAc)3 (1.78 eq., 5.87 mmol, 1.244 g) in dichloroethane (3 mL), followed by the addition of tert-butyl (4-aminobutyl) carbamate 17 (1 eq., 3.30 mmol, 0.621 g). The reaction was stirred at room temperature for 48 h. The reaction mixture was quenched with a 1N NaOH solution to obtain a pH ~ 8 of the aqueous layer. The reaction mixture was extracted three times with dichloromethane (3×) and the combined organic layers were concentrated to a volume of approximately 3 mL. Heptane (10 mL) was added and the volume was concentrated to 5 mL. Upon cooling the reaction mixture to room temperature, a precipitate was formed. After further cooling the suspension to 0 °C, the precipitate was filtered off and dried under vacuum, yielding the title compound as a light brown solid (674 mg, 64%); mp 73–75 °C. 1H NMR (400 MHz, CDCl3) δ: 8.39 (d, J = 4.1 Hz, 1H), 7.82 (s, 1H), 7.41 (d, J = 7.8 Hz, 1H), 7.12 (t, J = 7.4, 4.9 Hz, 1H), 4.01 (s, 1H), 3.09 (t, J = 33.0 Hz, 2H), 2.90–2.70 (m, 4H), 2.32–2.00 (m, 2H), 1.95 (t, J = 16.3 Hz, 2H), 1.73–1.62 (m, 2H), 1.62–1.51 (m, 2H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 156.31, 147.03, 137.27, 132.95, 122.48, 57.79, 46.09, 28.64, 27.77, 27.40, 26.52, 22.02, 20.18. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C18H29N3O2: 320.2332; found: 320.2335.
3.2.7. Coupling of 1-(Chloromethyl)isoquinolines 15a–d with Amine 18
General procedure
To a solution of 1-(chloromethyl)isoquinoline 15a–d (1.5 eq.) and tert-butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate 18 (1 eq.) in dry acetonitrile (5 mL) was added K2CO3 (8 eq.). The reaction mixture was refluxed for 24–48 h. The mixture was cooled to room temperature and filtered through a Celite® pad. The filtrate was evaporated in vacuo and the crude residue was purified via silica gel flash chromatography using a mixture of Et2O/MeOH as the mobile phase (in a gradient gradually ranging from 100:0 to 90:10) yielding the title compounds. The following compounds were made according to this procedure.
tert-Butyl(4-(((6,7-dimethoxyisoquinolin-1-yl)methyl)(5,6,7,8-tetrahydroquinolin-8 yl)amino)butyl) carbamate
This compound was prepared from 1-(chloromethyl)-6,7-dimethoxyisoquinoline 15a (86 mg, 0.36 mmol) and tert-butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate 18 (100 mg, 0.32 mmol). The title compound was isolated as a brown viscous oil (114 mg, 69%). 1H NMR (300 MHz, CDCl3) δ: 8.52 (s, 1H), 8.43 (d, J = 4.2 Hz, 1H), 8.26 (d, J = 5.6 Hz, 1H), 7.37 (d, J = 5.6 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 7.03–6.97 (m, 2H), 4.62 (s, 1H), 4.35 (dd, J = 56.5, 12.4 Hz, 2H), 4.14 (s, 3H), 4.00 (s, 3H), 2.97–2.46 (m, 6H), 2.12–1.90 (m, J = 22.3, 11.4 Hz, 3H), 1.70–1.54 (m, 1H), 1.39 (s, 9H), 1.34–1.16 (m, J = 28.5, 11.3 Hz, 4H).13C NMR (75 MHz, CDCl3) δ: 156.09, 152.72, 149.61, 147.10, 140.57, 136.49, 134.38, 133.31, 124.39, 121.55, 119.29, 106.62, 104.75, 77.36, 60.90, 58.81, 56.51, 55.99, 50.53, 29.55, 28.54, 27.74, 25.31, 24.94, 21.80. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C30H40N4O4: 521.3122; found: 521.3124.
tert-Butyl(4-(((6-methoxyisoquinolin-1-yl)methyl)(5,6,7,8-tetrahydroquinolin-8-yl) amino)butyl) carbamate
This compound was prepared from 1-(chloromethyl)-6-methoxyisoquinoline 15b (104 mg, 0.33 mmol) and tert-butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate 18 (101.4 mg, 0.49 mmol). The title compound was isolated as a brown viscous oil (136 mg, 85%). 1H NMR (400 MHz, CDCl3) δ: 8.66 (d, J = 9.2 Hz, 1H), 8.54 (d, J = 3.9 Hz, 1H), 8.32 (d, J = 5.8 Hz, 1H), 7.43 (d, J = 5.8 Hz, 1H), 7.36 (d, J = 7.4 Hz, 1H), 7.18 (dd, J = 9.3, 2.5 Hz, 1H), 7.08 (dd, J = 7.6, 4.6 Hz, 1H), 7.01 (d, J = 2.6 Hz, 1H), 4.89 (s, 1H), 4.38 (s, 1H), 4.24 (s, 2H), 3.94 (s, 3H), 2.94–2.56 (m, 6H), 2.42–2.12 (m, 4H), 2.15–1.99 (m, 4H), 1.39 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 161.5, 156.8, 147.8, 142.5, 139.1, 137.3, 135.2, 129.9, 122.5, 120.6, 120, 105, 56.1, 51.5, 40.5, 30.4, 30, 29.2, 28.2, 25.4, 22.2. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C29H38N4O3: 491.3016; found: 491.3017.
tert-Butyl(4-(((6-fluoroisoquinolin-1-yl)methyl)(5,6,7,8-tetrahydroquinolin-8-yl) amino)butyl) carbamate
This compound was prepared from 1-(chloromethyl)-6-fluoroisoquinoline 15c (117 mg, 0.6 mmol) and tert-butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate 18 (127.8 mg, 0.4 mmol). The title compound was isolated as a brown viscous oil (164 mg, 86%). 1H NMR (400 MHz, CDCl3) δ: 8.95 (dd, J = 8.9, 5.8 Hz, 1H), 8.51 (d, J = 3.9 Hz, 1H), 8.36 (d, J = 5.7 Hz, 1H), 7.47 (d, J = 5.7 Hz, 1H), 7.38–7.29 (m, 3H), 7.05 (dd, J = 7.6, 4.7 Hz, 1H), 4.80 (s, 1H), 4.36 (d, J = 12.5 Hz, 1H), 4.20 (d, J = 3.6 Hz, 2H), 2.95–2.52 (m, 6H), 2.14–1.97 (m, 2H), 1.88–1.63 (m, 4H), 1.39 (s, 9H), 1.30–1.22 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 155.7, 146.8, 141.9, 137.7, 137.6, 136.3, 134.2, 130.8, 124.9, 121.4, 119.8, 109.6, 109.3, 78.5, 60.8, 57.2, 53.2, 50.6, 39.7, 33.3, 29.4, 29, 28.1, 27.2, 24.5, 24, 21.2. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C28H35FN4O2: 479.2817; found: 479.2808.
tert-Butyl(4-(((6-bromoisoquinolin-1-yl)methyl)(5,6,7,8-tetrahydroquinolin-8-yl) amino)butyl) carbamate
This compound was prepared from 1-(chloromethyl)-6-bromoisoquinoline 15d (142 mg, 0.55 mmol) and tert-butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate 18 (117.9 mg, 0.37 mmol). The title compound was isolated as a brown viscous oil (143 mg, 72%).1H NMR (400 MHz, CDCl3) δ: 8.78 (d, J = 8.9 Hz, 1H), 8.53 (d, J = 3.8 Hz, 1H), 8.40 (d, J = 5.7 Hz, 1H), 7.92 (d, J = 1.9 Hz, 1H), 7.64 (dd, J = 9, 1.97 Hz, 1H), 7.43 (d, J = 5.7 Hz, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.06 (dd, J = 7.6, 4.69 Hz, 1H), 4.80 (s, 1H), 4.36 (d, J = 12, 1H), 4.20 (d, J = 10 Hz, 2H), 2.95–2.54 (m, 6H), 2.11–2.03 (m, 2H), 1.75–1.58 (m, 4H), 1.41 (s, 9H), 1.21 (t, J = 7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 155.9, 146.8, 141.9, 137.6, 137.3, 136.6, 130.2, 128.9, 128.7, 124.8, 123.2, 121.8, 119.3, 61.2, 57.1, 50.8, 39.6, 30.7, 29.3, 28.9, 28.2, 27.2, 27.5, 26.9, 24.3, 24, 21.2, 19.9. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C28H35BrN4O2: 539.2016; found: 539.2021.
This compound was prepared from the commercially available 1-(bromomethyl)isoquinoline (200 mg, 0.9 mmol) and tert-butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate 18 (191.78 mg, 0.6 mmol). The title compound was isolated as a brown viscous oil (169 mg, 61%). 1H NMR (400 MHz, CDCl3) δ: 8.52 (d, J = 3.8 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.68–7.61 (m, 1H), 7.52–7.43 (m, 1H), 7.33 (d, J = 7.5 Hz, 1H), 7.04 (dd, J = 7.6, 4.7 Hz, 1H), 4.86 (s, 1H), 4.21 (dd, J = 8.3, 6.6 Hz, 1H), 4.01–3.87 (m, 2H), 2.99 (s, 2H), 2.86–2.64 (m, 4H), 2.11–1.87 (m, 2H), 1.54–1.44 (m, 2H), 1.42 (s, 9H), 1.29–1.20 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 156.5, 147.7, 137.1, 136.6, 134.8, 129.6, 129.2, 128, 127.9, 126.3, 122.1, 121.9, 61.5, 58.9, 53.9, 52.8, 40.7, 30.2, 29.7, 28.9, 28.1, 26, 21.9. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C28H36N4O2: 461.2911; found: 461.2935.
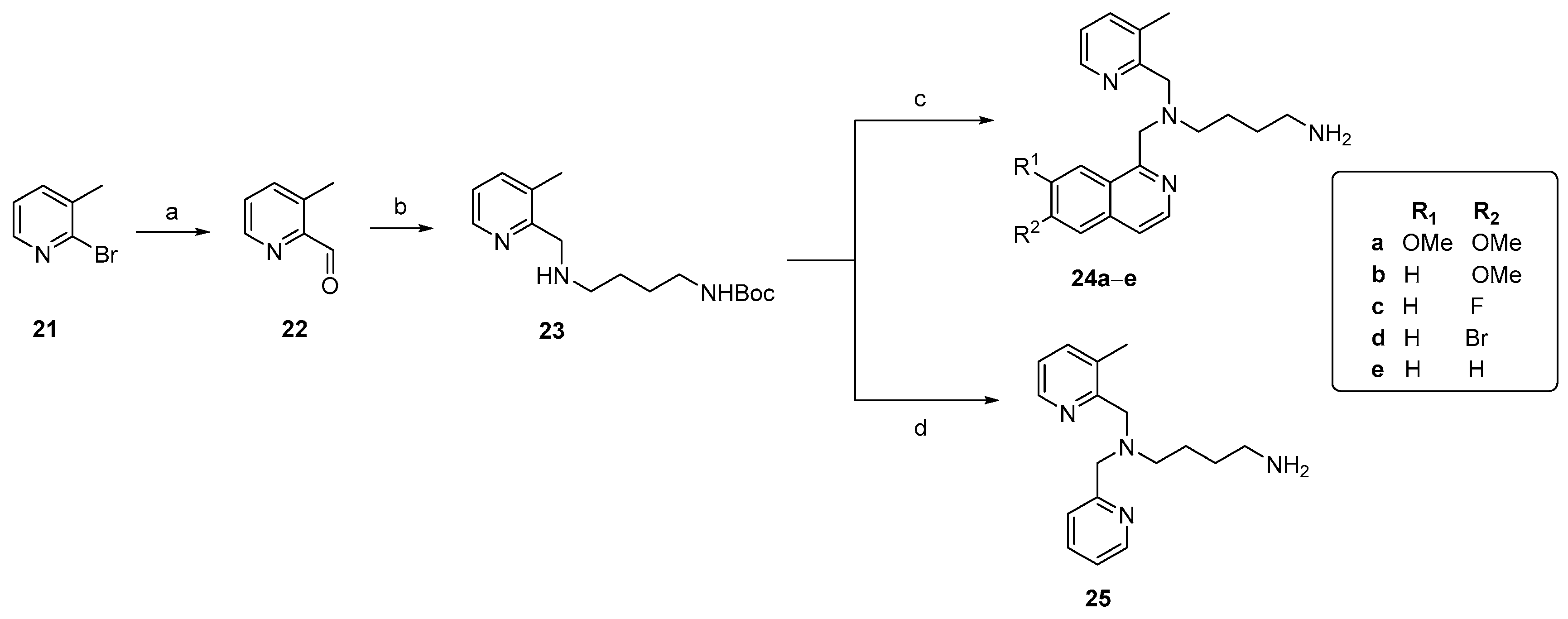
3.2.8. Synthesis of 3-Methylpicolinaldehyde (22)
To a flame-dried flask under an inert argon atmosphere was added 2-bromo-3-methyl-pyridine 21 (1 eq., 11.40 mmol, 2 g) and Et2O (1–2 mL) at −78°C, followed by the addition of a 1.6 M butyllithium solution in hexane (0.43 mL). After stirring for 3 h at −78 °C, DMF (2 mL) was added to this deep red-brown solution. The reaction temperature was gradually increased to room temperature and was stirred for an additional 22 h. The reaction mixture was quenched through the dropwise addition of ice-water and extracted with Et2O. The organic layer was washed with a 5% aqueous NaHCO3 solution and brine. The combined organic layers were dried over MgSO4 and concentrated under vacuum. The crude residue was purified by silica gel flash chromatography using Et2O/petroleum ether (80:20) as the eluent, yielding the title compound as an orange-red oil (848 mg, 62%).
3.2.9. tert-Butyl(4-(((3-methylpyridin-2-yl)methyl)amino)butyl) carbamate (23)
3-Methylpicolinaldehyde 22 (1 eq., 8.25 mmol, 1 g) was added to a slurry of NaBH(OAc)3 (1.9 eq., 15.68 mmol, 3.324 g) in dichloroethane (15 mL), followed by the addition of tert-butyl (4-aminobutyl) carbamate 17 (1.4 eq., 11.56 mmol, 2.176 g). The reaction was stirred at room temperature for 48 h. The reaction mixture was quenched with a 1 N NaOH solution to obtain a pH ~ 8 in the aqueous layer. After extracting the mixture with dichloromethane (3×), the combined organic phases were concentrated to a volume of approximately 3 mL. The crude residue was purified by silica gel column chromatography using Et2O/MeOH (96:4) as the eluent, affording the title compound as a yellowish viscous oil (1.8 g, 74%). 1H NMR (400 MHz, CDCl3) δ: 8.37 (d, J = 3.84 Hz, 1H), 7.41 (dd, J = 7.6, 0.64 Hz, 1H), 7.06 (dd, J = 7.5, 4.8 Hz, 1H), 4.79 (s, 1H), 3.86 (s, 2H), 3.17–3.06 (m, 2H), 2.94 (s, 1H), 2.72 (t, J = 6.7 Hz, 2H), 2.29 (s, 3H), 1.64–1.49 (m, 4H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 157.1, 156.1, 146.5, 137.7, 130.9, 121.9, 79, 52, 49.5, 40.5, 28.5, 27.4, 18.1. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C16H27N3O2: 294.2176; found: 294.2169.
3.2.10. Coupling of 1-(Chloromethyl)isoquinolines 15a–d with amine 23
A similar procedure as mentioned earlier for the coupling with amine 18 was used. The following compounds were made according to this procedure.
This compound was prepared from 1-(chloromethyl)-6,7-dimethoxyisoquinoline 15a (50 mg, 0.21 mmol) and tert-butyl (4-(((3-methylpyridin-2-yl)methyl)amino)butyl)carbamate 23 (48 mg, 0.15 mmol). The title compound was isolated as a brownish viscous oil (76 mg, 73%). 1H NMR (400 MHz, CDCl3) δ: 8.38 (d, J = 4.1 Hz, 1H), 8.30 (d, J = 5.6 Hz, 1H), 7.51 (s, 1H), 7.42 (d, J = 5.6 Hz, 1H), 7.36 (d, J = 7.4 Hz, 1H), 7.08 (dd, J = 7.6, 4.8 Hz, 1H), 7.02 (s, 1H), 4.75 (s, 1H), 4.23 (s, 2H), 4.00 (s, 3H), 3.82 (s, 5H), 2.96–2.83 (m, 2H), 2.59 (s, 2H), 2.10 (s, 3H), 1.64–1.51 (m, 2H), 1.40 (s, 9H), 1.30 (t, J = 6.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 162.5, 156.8, 155.9, 151.9, 151.3, 147.2, 141.2, 136.8, 133.1, 131.9, 123.9, 121.3, 117.7, 105.4, 79.5, 67.6, 59.1, 56.1, 54, 35.8, 28.4, 27.7, 25.8, 20.4. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C28H38N4O4: 495.2966; found: 495.2960.
This compound was prepared from 1-(chloromethyl)-6-methoxyisoquinoline 15b (100 mg, 0.48 mmol) and tert-butyl (4-(((3-methylpyridin-2-yl)methyl)amino)butyl)carbamate 23 (94.2 mg, 0.32 mmol). The title compound was isolated as a brownish viscous oil (118 mg, 57%). 1H NMR (400 MHz, CDCl3) δ: 8.38 (d, J = 3.8 Hz, 1H), 8.33 (d, J = 5.8 Hz, 1H), 7.88 (d, J = 9 Hz, 1H), 7.43 (d, J = 5.8 Hz, 1H), 7.40 (d, J = 7.1 Hz, 1H), 7.10 (dd, J = 7.5, 4.8 Hz, 1H), 7.06–6.98 (m, 2H), 4.71 (s, 1H), 4.14 (s, 2H), 3.91 (s, 3H), 3.83 (s, 2H), 2.94–2.82 (m, 2H), 2.57 (t, J = 7.2 Hz, 2H), 2.10 (s, 3H), 1.58–1.46 (m, 2H), 1.44 (s, 9H), 1.32–1.17 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 160.2, 155.8, 146, 141.8, 138.2, 137.8, 133.2, 128.1, 123.2, 122.3, 119.7, 119.1, 104.1, 78.7, 59.1, 55.2, 53.7, 39.7, 28.2, 27.4, 22.7, 18. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C27H36N4O3: 465.2860; found: 465.2863.
This compound was prepared from 1-(chloromethyl)-6-fluoroisoquinoline 15c (120 mg, 0.61 mmol) and tert-butyl (4-(((3-methylpyridin-2-yl)methyl)amino)butyl)carbamate 23 (120 mg, 0.41 mmol). The title compound was isolated as a brown viscous oil (150 mg, 81%). 1H NMR (400 MHz, CDCl3) δ: 8.41 (d, J = 5.7 Hz, 2H), 8.06 (dd, J = 9.2, 5.7 Hz, 1H), 7.49 (d, J = 5.8 Hz, 1H), 7.42 (d, J = 6.9 Hz, 1H), 7.35 (dd, J = 9.3, 2.5 Hz, 1H), 7.17 (dd, J = 8.8, 2.4 Hz, 1H), 7.12 (dd, J = 7.6, 4.9 Hz, 1H), 4.66 (s, 1H), 4.16 (s, 2H), 3.83 (s, 2H), 2.97–2.86 (m, 2H), 2.56 (t, J = 7.3 Hz, 2H), 2.13 (s, 3H), 1.59–1.48 (m, 2H), 1.42 (s, 9H), 1.31–1.19 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 164.1, 161.6, 159, 156.9, 156.1, 146.4, 142.4, 138, 133.3, 129.9, 125, 122.6, 120.2, 116.9, 110, 79, 59.6, 59.4, 54.1, 40, 28.5, 27.8, 23, 18.3. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C26H33FN4O2: 453.2660; found: 453.2651.
This compound was prepared from 1-(chloromethyl)-6-bromoisoquinoline 15d (100 mg, 0.39 mmol) and tert-butyl (4-(((3-methylpyridin-2-yl)methyl)amino)butyl)carbamate 23 (76.25 mg, 0.26 mmol). The title compound was isolated as a brown viscous oil (104 mg, 78%). 1H NMR (400 MHz, CDCl3) δ: 8.43 (d, J = 5.8 Hz, 1H), 8.40 (d, J = 3.9 Hz, 1H), 7.93 (d, J = 1.9 Hz, 1H), 7.85 (d, J = 9 Hz, 1H), 7.50–7.41 (m, 3H), 7.12 (dd, J = 7.6, 4.8 Hz, 1H), 4.65 (s, 1H), 4.15 (s, 2H), 3.83 (s, 2H), 2.96–2.87 (m, 2H), 2.56 (t, J = 7.3 Hz, 2H), 2.16 (s, 3H), 1.75–1.60 (m, 2H), 1.58–1.47 (m, 2H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 157.1, 156.4, 146.7, 142.8, 138.4, 137.9, 133.7, 130.4, 129.3, 128.7, 126.5, 125, 123, 119.9, 79.3, 59.7, 54.5, 40.3, 28.8, 28.1, 23.3, 18.7. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C26H33BrN4O2: 513.1860; found: 513.1854.
This compound was prepared from 1-(bromomethyl)isoquinoline (100 mg, 0.45 mmol) and tert-butyl (4-(((3-methylpyridin-2-yl)methyl)amino)butyl)carbamate 15 (88 mg, 0.3 mmol). The title compound was isolated as a brown viscous oil (110 mg, 84%). 1H NMR (400 MHz, CDCl3) δ: 8.37 (dd, J = 4.7, 1.1 Hz, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.77 (dd, J = 8.2, 1.3 Hz, 1H), 7.71–7.65 (m, 1H), 7.54–7.47 (m, 2H), 7.39 (dd, J = 7.7, 0.7 Hz, 1H), 7.06 (dd, J = 7.6, 4.8 Hz, 1H), 4.74 (s, 1H), 3.91 (s, 2H), 3.86 (s, 2H), 3.06–2.93 (m, 2H), 2.57 (t, J = 7.2 Hz, 2H), 2.33 (s, 3H), 1.70–1.61 (m, 2H), 1.59–1.50 (m, 2H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 161.2, 157.5, 156.4, 147.9, 146.6, 138.4, 136.4, 133.6, 129.7, 129.5, 127.9, 127.7, 126.5, 122.8, 121.9, 61.8, 60.1, 54.7, 40.6, 28.9, 28.2, 24.1, 19. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C26H34N4O2: 435.2754; found: 435.2757.
3.2.11. General Procedure for Deprotection of the Basic n-Butylamine Side Chain from Boc-Protected Intermediates
General procedure
To a solution of the Boc-protected intermediate (0.1 mmol) in dry dichloromethane (1 mL) was added trifluoroacetic acid (0.3 mL). The reaction mixture was stirred for 1 h at room temperature. The solvents were evaporated in vacuo. The residue was neutralized by adding a saturated aqueous NaHCO3 solution and the aqueous layer was extracted five times with dichloromethane. The combined organic layers were dried over MgSO4 and evaporated. The crude residue was purified via silica gel flash column chromatography using a mixture of DCM/MeOH (in a ratio gradually ranging from 95:5 to 90:10) as the mobile phase. The following compounds were made according to this procedure.
N1-((6,7-Dimethoxyisoquinolin-1-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine (19a)
This compound was prepared from its Boc-protected precursor (50 mg, 0.09 mmol). The title compound was isolated as a bright brown viscous oil (47 mg, 95%). 1H NMR (400 MHz, DMSO) δ (ppm): 8.43 (d, J = 4.5 Hz, 1H), 8.21 (d, J = 5.6 Hz, 1H), 8.17 (s, 1H), 7.55 (d, J = 5.4 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.30 (s, 1H), 7.20–7.10 (m, 1H), 5.32 (s, 1H), 4.29 (dd, J = 21.5, 12.0 Hz, 2H), 3.99 (s, 3H), 3.92 (s, 3H), 2.15–1.50 (m, 6H), 1.39 (s, 2H), 1.30–1.09 (m, 4H), 1.07–0.93 (m, J = 8.3, 6.1 Hz, 4H). 13C NMR (101 MHz, DMSO) δ: 158.51, 158.20, 157.89, 157.59, 152.18, 139.94, 132.61, 123.25, 121.82, 121.65, 119.06, 118.84, 116.52, 115.85, 112.78, 55.51, 52.06, 49.46, 48.60, 45.67, 43.69, 7.17. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C25H32N4O2: 421.2598; found: 421.2594.
N1-((6-Methoxyisoquinolin-1-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine (19b)
This compound was prepared from its Boc-protected precursor (30 mg, 0.06 mmol). The title compound was isolated as a bright brown viscous oil (22 mg, 92%). 1H NMR (400 MHz, CDCl3) δ: 9.99 (s, 2H), 8.54 (d, J = 3.7 Hz, 1H), 8.51 (d, J = 5.8 Hz, 1H), 8.03 (d, J = 9.3 Hz, 1H), 7.51 (d, J = 5.8 Hz, 1H), 7.41 (d, J = 7.2 Hz, 1H), 7.22 (dd, J = 9.3, 2.5 Hz, 1H), 7.12 (dd, J = 7.7, 4.8 Hz, 1H), 7.08 (d, J = 2.5 Hz, 1H), 4.47 (d, J = 15 Hz, 1H), 4.05 (d, J = 15 Hz, 2H), 3.07 (s, 3H), 3.01–2.46 (m, 6H), 2.12–1.91 (m, 4H), 1.81–1.54 (m, 4H). 13C NMR (101 MHz, CDCl3) δ: 161.1, 156.6, 156, 146.9, 141.6, 139, 138, 135, 125.9, 123, 122.7, 120.9, 120.4, 105.3, 60.2, 55.7, 51.6, 51.3, 39.7, 31.1, 29.3, 27.7, 26.9, 21.7, 21.12. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C24H30N4O: 391.2492; found: 391.2487.
N1-((6-Fluoroisoquinolin-1-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine (19c)
This compound was prepared from its Boc-protected precursor (27 mg, 0.06 mmol). The title compound was isolated as a bright brown viscous oil (21 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 9.84 (s, 2H), 8.60 (d, J = 5.8 Hz, 1H), 8.52 (d, J = 3.5 Hz, 1H), 8.19 (dd, J = 9.3, 5.3 Hz, 1H), 7.57 (d, J = 5.8 Hz, 1H), 7.47–7.32 (m, 3H), 7.11 (dd, J = 7.6, 4.8 Hz, 1H), 4.46 (d, J = 15.1 Hz, 1H), 4.11 (d, J = 14.9 Hz, 2H), 2.98–2.52 (m, 6H), 2.12–1.94 (m, 4H), 1.82–1.58 (m, 4H). 13C NMR (101 MHz, CDCl3) δ: 164.5, 162, 157.3, 155.8, 147, 142.2, 138.2, 135, 127.4, 124.5, 122.8, 120.7, 118.3, 111.2, 60.3, 52.2, 51.5, 39.6, 29.8, 29.4, 27.7, 26.6, 21.7, 21.2. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C23H27FN4: 379.2292; found: 379.2286.
N1-((6-Bromoisoquinolin-1-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine (19d)
This compound was prepared from its Boc-protected precursor (22 mg, 0.04 mmol). The title compound was isolated as a bright brown viscous oil (18 mg, 99%). 1H NMR (600 MHz, CDCl3) δ: 8.63 (d, J = 5.8 Hz, 1H), 8.54 (d, J = 3.7 Hz, 1H), 8.18 (d, J = 8.9 Hz, 1H), 8.01 (d, J = 1.8 Hz, 1H), 7.68 (dd, J = 9, 1.9 Hz, 1H), 7.52 (d, J = 5.8 Hz, 1H), 7.41 (d, J = 7.2 Hz, 1H), 7.12 (dd, J = 7.7, 4.7 Hz, 1H), 4.44 (d, J = 14.8 Hz, 1H), 4.12 (d, J = 14.9 Hz, 2H), 2.91–2.51 (m, 6H), 2.12–1.84 (m, 4H), 1.74–1.54 (m, 4H). 13C NMR (151 MHz, CDCl3) δ: 162.4, 158, 147.3, 14306, 136.8, 134.5, 132, 129.7, 128.4, 127.9, 125.5, 123.3, 121.3, 118.1, 67.7, 59.2, 54, 41.8, 28.5, 26.3, 25.8, 20.5. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C23H27BrN4: 439.1492; found: 439.1495.
N1-(Isoquinolin-1-ylmethyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine (19e)
This compound was prepared from its Boc-protected precursor (21 mg, 0.05 mmol). The title compound was isolated as a bright brown viscous oil (16 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 10.04 (s, 2H), 8.61 (d, J = 3.7 Hz, 1H), 8.20 (d, J = 8.7 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 7.7 Hz, 2H), 7.58–7.51 (m, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.13 (dd, J = 7.6, 4.8 Hz, 1H), 4.06 (d, J = 14.2 Hz, 2H), 3.92 (d, J = 14.2 Hz, 1H), 3.03–2.67 (m, 6H), 2.13–1.89 (m, 4H), 1.76–1.53 (m, 4H). 13C NMR (101 MHz, CDCl3) δ: 162.4, 158.3, 147.3, 142.8, 136.8, 136.5, 132, 129.7, 127.5, 127.3, 126.8, 124.3, 121.3, 119.2, 67.7, 59.2, 54, 41.8, 28.5, 26.3, 25.8, 20.5. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C23H28N4: 361.2387; found: 361.2376.
This compound was prepared from its Boc-protected precursor (17 mg, 0.03 mmol). The title compound was isolated as a bright brown viscous oil (13 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 8.52 (d, J = 4Hz, 1H), 8.43 (d, J = 5.6 Hz, 1H), 7.47 (d, J = 5.6 Hz, 1H), 7.44 (d, J = 6.9 Hz, 1H), 7.27 (s, 1H), 7.12 (dd, J = 7.7, 4.9 Hz, 1H), 7.07 (s, 1H), 4.20 (s, 2H), 4.02 (s, 3H), 4.00 (s, 3H), 3.83 (s, 2H), 3.15 (t, J = 5.4 Hz, 2H), 2.69 (t, J = 5.3 Hz, 2H), 2.26 (s, 3H), 1.89–1.84 (m, 2H), 1.82–1.77 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 155.3, 153.9, 153, 150.4, 146.7, 140, 138.6, 1336, 131.8, 122.8, 122.6, 119.6, 105.4, 102.3, 57.1, 56.6, 561, 55.2, 39.5, 30.9, 29.6, 27.3, 26.2, 18.5. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C23H30N4O2: 395.2441; found: 395.2434.
This compound was prepared from its Boc-protected precursor (22 mg, 0.05 mmol). The title compound was isolated as a bright brown viscous oil (17 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 8.46 (dd, J = 4.8, 1.1 Hz, 1H), 8.41 (d, J = 5.8 Hz, 1H), 7.96 (d, J = 9.3 Hz, 1H), 7.45 (d, J = 5.8 Hz, 1H), 7.41 (dd, J = 7.7, 0.7 Hz, 1H), 7.15 (dd, J = 9.3, 2.6 Hz, 1H), 7.09 (dd, J = 7.6, 4.8 Hz, 1H), 7.03 (d, J = 2.5 Hz, 1H), 4.13 (s, 2H), 3.92 (s, 3H), 3.78 (s, 2H), 2.99–2.89 (m, 2H), 2.59 (t, J = 5.7 Hz, 2H), 2.20 (s, 3H), 1.76–1.63 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 160.8, 156.9, 155.9, 146.7, 142, 138.7, 132.3, 126.6, 123, 122.7, 120.4, 120.2, 105, 57.7, 57.3, 55.6, 55.2, 39.8, 31.1, 28, 25.6, 18.6. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C22H28N4O: 365.2336; found: 365.2330.
This compound was prepared from its Boc-protected precursor (28 mg, 0.062 mmol). The title compound was isolated as a bright brown viscous oil (21.5 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 8.51 (d, J = 5.8 Hz, 1H), 8.45 (d, J = 3.8 Hz, 1H), 8.12 (dd, J = 9.3, 5.4 Hz, 1H), 7.52 (d, J = 5.8 Hz, 1H), 7.44–7.36 (m, 2H), 7.32 (td, J = 8.9, 2.5 Hz, 1H), 7.08 (dd, J = 7.6, 4.8 Hz, 1H), 4.22 (s, 2H), 3.82 (s, 2H), 3.01 (t, J = 5.3 Hz, 2H), 2.69 (t, J = 5.2 Hz, 2H), 2.22 (s, 3H), 1.86–1.69 (m, 4H). 13C NMR (101 MHz, CDCl3) δ: 163.9, 161.4, 158, 156, 146.3, 142.2, 137.9, 132.5, 128.7, 124.5, 122.4, 120.1, 117.1, 110.2, 58.3, 54.7, 40.5, 30.7, 29.5, 24.2, 18.2. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C21H25FN4: 353.2136; found: 353.2131.
This compound was prepared from its Boc-protected precursor (22 mg, 0.043 mmol). The title compound was isolated as a bright brown viscous oil (17.6 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 8.57 (d, J = 5.7 Hz, 1H), 8.46 (d, J = 3.9 Hz, 1H), 7.97 (d, J = 1.8 Hz, 1H), 7.94 (d, J = 9.1 Hz, 1H), 7.63 (dd, J = 9, 1.9 Hz, 1H), 7.47 (d, J = 5.8 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.09 (dd, J = 7.6, 4.8 Hz, 1H), 4.19 (s, 2H), 3.82 (s, 2H), 3.04 (t, J = 4.7 Hz, 2H), 2.68 (t, J = 5.5 Hz, 2H), 2.22 (s, 3H), 1.87–1.70 (m, 4H). 13C NMR (101 MHz, CDCl3) δ: 157.3, 155.2, 146.5, 142.3, 138.4, 137.3, 131.9, 130.8, 129.5, 126.1, 125.4, 125, 122.4, 119.5, 56.9, 55, 39.3, 30.8, 29.5, 27.1, 25.3, 18.4. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C21H25BrN4: 413.1336; found: 413.1339.
This compound was prepared from its Boc-protected precursor (22 mg, 0.05 mmol). The title compound was isolated as a bright brown viscous oil (17 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 8.43 (dd, J = 4.8, 1.1 Hz, 1H), 8.08 (dd, J = 8.3, 4.7 Hz, 2H), 7.77 (dd, J = 8.1, 1.1 Hz, 1H), 7.74–7.68 (m, 1H), 7.54–7.49 (m, 1H), 7.47 (d, J = 8.5 Hz, 1H), 7.41 (dd, J = 7.6, 0.9 Hz, 1H), 7.08 (dd, J = 7.6, 4.8 Hz, 1H), 3.92 (s, 2H), 3.85 (s, 2H), 2.75 (t, J = 6.5 Hz, 2H), 2.58 (t, J = 6.8 Hz, 2H), 2.35 (s, 3H), 1.65–1.56 (m, 2H), 1.53–1.43 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 158.1, 155.1, 147.2, 146.4, 138.4, 137.1, 131.7, 130.2, 128.2, 127.4, 127.1, 126.5, 122.4, 122.3, 61, 56.9, 55, 39.2, 30.7, 29.5, 26.9, 25.6, 18.3. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C21H26N4: 335.2230; found: 335.2228.
3.2.12. tert-Butyl (4-((pyridin-2-ylmethyl)(5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate
Picolinaldehyde (1 eq., 0.6887 mmol, 0.0655 mL) was added to a slurry of NaBH(OAc)3 (1.78 eq., 1.2259 mmol, 259.8 mg) in dichloromethane (3 mL), followed by the addition of tert-butyl (4-((5,6,7,8-tetrahydroquinolin-8-yl)amino)butyl)carbamate 18 (1 eq., 0.6887 mmol, 220 mg). The reaction mixture was stirred at room temperature for 48 h. Then, the reaction was quenched with a 1N NaOH solution to obtain pH ~ 8 of the aqueous layer. The aqueous layer was extracted three times with dichloromethane. The combined organic phases were concentrated and the crude residue was purified via silica gel flash chromatography using a mixture of DCM/MeOH (in a ratio of 96:4) as mobile phase, affording the title compound as a brown viscous oil (177 mg, 63%). 1H NMR (400 MHz, CDCl3) δ: 8.49 (d, J = 3.7 Hz, 1H), 8.45 (d, J = 4.8 Hz, 1H), 7.73 (d, J = 7.9 Hz, 1H), 7.63, (td, J = 7.5, 1.7 Hz, 1H), 7.33 (d, J = 7.5 Hz, 1H), 7.10 (dd, J = 6.5, 5.6 Hz, 1H), 7.04 (dd, J = 7.6, 4.7 Hz, 1H), 4.8 (s, 1H), 4.13 (d, J = 7.4 Hz, 1H), 3.91 (d, J = 14.6 Hz, 2H), 3.09–2.94 (m, 2H), 2.86–2.63 (m, 4H), 2.22–2.09 (m, 2H), 2.06–1.95 (m, 2H), 1.94–1.81 (m, 2H), 1.76–1.61 (m, 2H), 1.41 (s, 9H).13C NMR (101 MHz, CDCl3) δ: 162.4, 157.9, 155.9, 148.7, 147.3, 139.7, 136.7, 132, 124.2, 121.2, 121, 79.5, 67.7, 60.9, 54, 35.9, 28.4, 27.7, 25.8, 20.5. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C24H34N4O2: 411.2754; found: 411.2757.
3.2.13. tert-Butyl (4-(((3-methylpyridin-2-yl)methyl)(pyridin-2-ylmethyl)amino)butyl)carbamate
Picolinaldehyde (1 eq., 1.02 mmol, 0.1 mL) was added to a slurry of NaBH(OAc)3 (1,78 eq., 1.82 mmol, 385.7 mg) in DCM (3 mL), followed by the addition of tert-butyl (4-(((3-methylpyridin-2yl)methyl)amino)butyl)carbamate 23 (1 eq., 1.02 mmol, 200 mg). The reaction was stirred at room temperature for 48 h. After completion, the reaction mixture was quenched utilizing 1 N NaOH solution to obtain pH ~ 8 in the aqueous layer. After extracting the mixture with DCM for 3 times, the combined organic phases were concentrated. Thereafter, the residue was purified through column chromatography using a DCM/MeOH (96:4) gradient elution to afford the corresponding compound. Brown viscous oil; yield: 206 mg (52%).1H NMR (400 MHz, CDCl3) δ: 8.50 (d, J = 4.1 Hz, 1H), 8.36 (dd, J = 4.8, 1.2 Hz, 1H), 7.59 (td, J = 7.7, 1.8 Hz, 1H), 7.39 (d, J = 6.7 Hz, 1H), 7.34 (d, J = 10.3 Hz, 1H), 7.12 (dddd, J = 7.4, 5, 1.1 Hz, 1H), 7.07 (dd, J = 7.6, 4.8 Hz, 1H), 4.81 (s, 1H), 3.80 (s, 2H), 3.75 (s, 2H), 3.06–2.92 (m, 2H), 2.53 (t, J = 7.2 Hz, 2H), 2.30 (s, 3H), 1.57–1.47 (m, 2H), 1.43 (s, 9H), 1.41–1.31 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 160.1, 157.3, 156.4, 149.1, 146.4, 138.3, 136.5, 133.5, 123.7, 122.7, 122.1, 60.7, 59.9, 54.4, 40.4, 28.8, 28.1, 24, 18.7. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C22H32N4O2: 385.2598; found: 385.2591.
3.2.14. N1-(Pyridin-2-ylmethyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine (20)
This compound was synthesized starting from its Boc-protected precursor (23 mg, 0.06 mmol), according to the general deprotection procedure, yielding the title compound as a brownish viscous oil (17 mg, 99%). 1H NMR (600 MHz, CDCl3) δ: 9.91 (s, 2H), 8.73 (dd, J = 4.9, 0.86 Hz, 1H), 8.58 (d, J = 3.9 Hz, 1H), 7.68 (td, J = 7.7, 1.8 Hz, 1H), 7.41 (d, J = 7 Hz, 1H), 7.29–7.21 (m, 2H), 7.15 (dd, J = 7.7, 4.8 Hz, 1H), 3.92 (d, J = 13.7 Hz, 2H), 3.73 (d, J = 13.7 Hz, 1H), 2.99–2.64 (m, 4H), 2.39–2.20 (m, 2H), 2.11–1.92 (m, 2H), 1.75–1.56 (m, 6H). 13C NMR (151 MHz, CDCl3) δ: 162.5, 157.9, 148.7, 17.2, 139.8, 136.7, 131.9, 124.2, 121.3, 121, 67.7, 60.9, 53.9, 41.8, 28.4, 26.3, 25.8, 20.5. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C19H26N4: 311.2230; found: 311.2226.
3.2.15. N1-((3-Methylpyridin-2-yl)methyl)-N1-(pyridin-2-ylmethyl)butane-1,4-diamine (25)
This compound was synthesized starting from its Boc-protected precursor (30 mg, 0.08 mmol), according to the general deprotection procedure, yielding the title compound as a brownish viscous oil (18 mg, 81%). 1H NMR (600 MHz, CDCl3) δ: 9.36 (s, 2H), 8.66 (d, J = 4.5 Hz, 1H), 8.52 (d, J = 4.5 Hz, 1H), 7.65 (td, J = 7.7, 1.6 Hz, 1H), 7.43 (d, J = 7.5 Hz, 1H), 7.21 (d, J = 7.4 Hz, 1H), 7.20 (d, J = 5.1 Hz, 1H), 7.12 (dd, J = 7.5, 4.9 Hz, 1H), 3.77 (s, 2H), 3.73 (s, 2H), 3.13 (t, J = 5.4 Hz, 2H), 2.58 (t, J = 5.2 Hz, 2H), 2.25 (s, 3H), 1.85–1.79 (m, 2H), 1.78–1.73 (m, 2H). 13C NMR (151 MHz, CDCl3) δ: 157.4, 1552, 149.8, 146.8, 138.9, 137.4, 132, 124, 123.1, 122.8, 60.4, 57, 55.1, 39.6, 29.8, 27.3, 262, 18.6. HRMS (ESI-Q-TOF): m/z [M + H]+ calcd for C17H24N4: 285.2074; found: 285.2077.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}