
Modulating Glycoside Hydrolase Activity between Hydrolysis and Transfer Reactions Using an Evolutionary Approach


Abstract
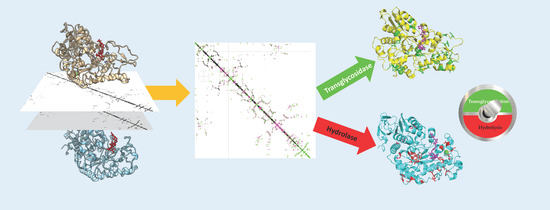
:
1. Introduction
2. Results
2.1. Homology Model of TmAmyA
2.2. Protein Classification as Hydrolases or Transglycosidases
2.3. Modification of Reaction Specificity in the α-Amylase A from Termotoga maritma
2.4. Increasing Hydrolase Activity in the 1,4-α-Glucanotransferase from Thermotoga maritima
2.5. Molecular Dynamic Simulations
3. Discussion
4. Materials and Methods
4.1. Bioinformatic Analysis
4.1.1. Analysis 1
4.1.2. Analysis 2
4.1.3. Analysis 3
4.1.4. Selection of Mutation Sites
4.1.5. Molecular Dynamic Simulations
4.1.6. Homology Models
4.2. Experimental
4.2.1. Construction of the TmGTase Gene Vector
4.2.2. Construction of TmAmyA and TmGTase Variants
4.2.3. TmAmyA and TmGTase Variants Expression
4.2.4. Characterization of TmAmyA Variants
4.2.5. Characterization of TmGTase Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Samples Availability
References
- Chapman, J.; Ismail, A.E.; Dinu, C.Z. Industrial applications of enzymes: Recent advances, techniques, and outlooks. Catalysts 2018, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.A.; Ali, S.; Hassan, A.; Tahir, H.M.; Mumtaz, S.; Mumtaz, S. Biosynthesis and industrial applications of α-amylase: A review. Arch. Microbiol. 2021, 203, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Prim. 2021, 1, 46. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. Streamlining design, engineering, and applications of enzymes for sustainable biocatalysis. ACS Sustain. Chem. Eng. 2021, 9, 8032–8052. [Google Scholar] [CrossRef]
- Gargiulo, S.; Soumillion, P. Directed evolution for enzyme development in biocatalysis. Curr. Opin. Chem. Biol. 2021, 61, 107–113. [Google Scholar] [CrossRef]
- de Pina Mariz, B.; Carvalho, S.; Batalha, I.L.; Pina, A.S. Artificial enzymes bringing together computational design and directed evolution. Org. Biomol. Chem. 2021, 19, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, M.J.; Leaver-Fay, A.; Tyka, M.D.; Stein, A.; Houlihan, K.; Dimaio, F.; Bradley, P.; Kortemme, T.; Baker, D.; Snoeyink, J.; et al. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 2015, 11, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Bradley, P.; Greisen, P.; Liu, Y.; Mulligan, V.K.; Kim, D.E.; Baker, D.; Dimaio, F. Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J. Chem. Theory Comput. 2016, 12, 6201–6212. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.; Ertelt, M.; Merkl, R.; Meiler, J. Rosetta design with co-evolutionary information retains protein function. PLoS Comput. Biol. 2021, 17, e1008568. [Google Scholar] [CrossRef]
- Petrovic, D.; Risso, V.A.; Kamerlin, S.C.L.; Sanchez-Ruiz, J.M. Conformational dynamics and enzyme evolution. J. R. Soc. Interface 2018, 15, 20180330. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Crean, R.M.; Gardner, J.M.; Kamerlin, S.C.L. Harnessing Conformational Plasticity to Generate Designer Enzymes. J. Am. Chem. Soc. 2020, 142, 11324–11342. [Google Scholar] [CrossRef] [PubMed]
- Bunzel, H.A.; Anderson, J.L.R.; Mulholland, A.J. Designing better enzymes: Insights from directed evolution. Curr. Opin. Struct. Biol. 2021, 67, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.D.; Beatty, J.T.; Landweber, L.F. The TIM Barrel Architecture Facilitated the Early Evolution of Protein-Mediated Metabolism. J. Mol. Evol. 2016, 82, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.L. Adaptive evolution after gene duplication. Trends Genet. 2002, 18, 433–434. [Google Scholar] [CrossRef]
- Hughes, A.L. The evolution of functionally novel proteins after gene duplication. Proc. R. Soc. Lon. B 1994, 256, 119–124. [Google Scholar] [CrossRef]
- Wierenga, R.K. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett. 2001, 492, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Schmiedel, J.M.; Lehner, B. Determining protein structures using deep mutagenesis. Nat. Genet. 2019, 51, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Rollins, N.J.; Brock, K.P.; Poelwijk, F.J.; Stiffler, M.A.; Gauthier, N.P.; Sander, C.; Marks, D.S. Inferring protein 3D structure from deep mutation scans. Nat. Genet. 2019, 51, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Salinas, V.H.; Ranganathan, R. Coevolution-based inference of amino acid interactions underlying protein function. Elife 2018, 7, 1–20. [Google Scholar] [CrossRef]
- Baussand, J.; Carbone, A. A combinatorial approach to detect coevolved amino acid networks in protein families of variable divergence. PLoS Comput. Biol. 2009, 5, e1000488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovchinnikov, S.; Kim, D.E.; Wang, R.Y.R.; Liu, Y.; Dimaio, F.; Baker, D. Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta. Proteins Struct. Funct. Bioinform. 2016, 84, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, D.; Huan, J.; Prins, J.; Snoeyink, J.; Wang, W.; Tropsha, A. Identification of family-specific residue packing motifs and their use for structure-based protein function prediction: II. Case studies and applications. J. Comput. Aided. Mol. Des. 2009, 23, 785–797. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Huan, J.; Prins, J.; Snoeyink, J.; Wang, W.; Tropsha, A. Identification of family-specific residue packing motifs and their use for structure-based protein function prediction: I. Method development. J. Comput. Aided. Mol. Des. 2009, 23, 773–784. [Google Scholar] [CrossRef]
- Yeang, C.H.; Haussler, D. Detecting coevolution in and among protein domains. PLoS Comput. Biol. 2007, 3, 2122–2134. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Figliuzzi, M.; Stocker, C.; Barrat-Charlaix, P.; Socolich, M.; Kast, P.; Hilvert, D.; Monasson, R.; Cocco, S.; Weigt, M.; et al. An evolution-based model for designing chorismate mutase enzymes. Science 2020, 369, 440–445. [Google Scholar] [CrossRef]
- Wang, X.; Jing, X.; Deng, Y.; Nie, Y.; Xu, F.; Xu, Y.; Zhao, Y.; Hunt, J.F.; Montelione, G.T.; Szyperski, T. Evolutionary coupling saturation mutagenesis: Coevolution-guided identification of distant sites influencing Bacillus naganoensis pullulanase activity. FEBS Lett. 2020, 594, 799–812. [Google Scholar] [CrossRef]
- Tondnevis, F.; Dudenhausen, E.E.; Miller, A.M.; McKenna, R.; Altschul, S.F.; Bloom, L.B.; Neuwald, A.F. Deep Analysis of Residue Constraints (DARC): Identifying determinants of protein functional specificity. Sci. Rep. 2020, 10, 1691. [Google Scholar] [CrossRef]
- Glycogenomics Group at AFMB Carbohydrate-Active enZYmes Database. Available online: http://www.cazy.org/ (accessed on 25 October 2019).
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, M.S.; Svensson, B. Structural biology of starch-degrading enzymes and their regulation. Curr. Opin. Struct. Biol. 2016, 40, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uitdehaag, J.C.; Mosi, R.; Kalk, K.H.; van der Veen, B.A.; Dijkhuizen, L.; Withers, S.G.; Dijkstra, B.W. X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the alpha-amylase family. Nat. Struct. Biol. 1999, 6, 432–436. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.L.; Cha, H.J.; Lee, J.S.; Park, S.H.; Woo, E.J.; Park, K.H. Introducing transglycosylation activity in Bacillus licheniformis α-amylase by replacement of His235 with Glu. Biochem. Biophys. Res. Commun. 2014, 451, 541–547. [Google Scholar] [CrossRef]
- Liebl, W.; Stemplinger, I.; Ruile, P. Properties and gene structure of the Thermotoga maritima alpha-amylase AmyA, a putative lipoprotein of a hyperthermophilic bacterium. J. Bacteriol. 1997, 179, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Roujeinikova, A.; Raasch, C.; Sedelnikova, S.; Liebl, W.; Rice, D.W. Crystal structure of Thermotoga maritima 4-α-glucanotransferase and its acarbose complex: Implications for substrate specificity and catalysis. J. Mol. Biol. 2002, 321, 149–162. [Google Scholar] [CrossRef]
- Blakeney, A.B.; Stone, B.A. Activity and action pattern of Bacillus licheniformis α-amylase in aqueous ethanol. FEBS Lett. 1985, 186, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Kuriki, T.; Kaneko, H.; Yanase, M.; Takata, H.; Shimada, J.; Handa, S.; Takada, T.; Umeyama, H.; Okada, S. Controlling substrate preference and transglycosylation activity of neopullulanase by manipulating steric constraint and hydrophobicity in active center. J. Biol. Chem. 1996, 271, 17321–17329. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.-J.; Yoon, H.-G.; Kim, Y.-W.; Lee, H.-S.; Kim, J.-W.; Kweon, K.-S.; Oh, B.-H.; Park, K.-H. Molecular and enzymatic characterization of a maltogenic amylase that hydrolyzes and transglycosylates acarbose. Eur. J. Biochem. 1998, 253, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geronimo, I.; Payne, C.M.; Sandgren, M. The role of catalytic residue pKa on the hydrolysis/transglycosylation partition in family 3 β-glucosidases. Org. Biomol. Chem. 2018, 16, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, T.; Hayashi, S. Crucial Role of Protein Flexibility in Formation of a Stable Reaction Transition State in an α-Amylase Catalysis. J. Am. Chem. Soc. 2012, 134, 7045–7055. [Google Scholar] [CrossRef] [PubMed]
- Damián-Almazo, J.Y.; Moreno, A.; López-Munguía, A.; Soberón, X.; González-Muñoz, F.; Saab-Rincón, G. Enhancement of the alcoholytic activity of α-amylase AmyA from Thermotoga maritima MSB8 (DSM 3109) by site-directed mutagenesis. Appl. Environ. Microbiol. 2008, 74, 5168–5177. [Google Scholar] [CrossRef] [Green Version]
- Rivera, M.H.; López-Munguía, A.; Soberón, X.; Saab-Rincón, G. A-Amylase from Bacillus licheniformis mutants near to the catalytic site: Effects on hydrolytic and transglycosylation activity. Protein Eng. 2003, 16, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saab-Rincón, G.; Del-Río, G.; Santamaría, R.I.; López-Munguía, A.; Soberón, X. Introducing transglycosylation activity in a liquefying α-amylase. FEBS Lett. 1999, 453, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Teze, D.; Hendrickx, J.; Dion, M.; Tellier, C.; Woods, V.L.; Tran, V.; Sanejouand, Y.H. Conserved water molecules in family 1 glycosidases: A DXMS and molecular dynamics study. Biochemistry 2013, 52, 5900–5910. [Google Scholar] [CrossRef]
- David, B.; Arnaud, P.; Tellier, C.; Sanejouand, Y.-H. Toward the design of efficient transglycosidases: The case of the GH1 of Thermus thermophilus. Protein Eng. Des. Sel. 2019, 32, 309–316. [Google Scholar] [CrossRef]
- Lundemo, P.; Karlsson, E.N.; Adlercreutz, P. Eliminating hydrolytic activity without affecting the transglycosylation of a GH1 β-glucosidase. Appl. Microbiol. Biotechnol. 2017, 101, 1121–1131. [Google Scholar] [CrossRef] [Green Version]
- Tran, L.T.; Blay, V.; Luang, S.; Eurtivong, C.; Choknud, S.; González-Díaz, H.; Ketudat Cairns, J.R. Engineering faster transglycosidases and their acceptor specificity. Green Chem. 2019, 21, 2823–2836. [Google Scholar] [CrossRef]
- Kelly, R.M.; Leemhuis, H.; Dijkhuizen, L. Conversion of a Cyclodextrin Glucanotransferase into an α-Amylase: Assessment of Directed Evolution Strategies. Biochemistry 2007, 46, 11216–11222. [Google Scholar] [CrossRef] [Green Version]
- Leemhuis, H.; Rozeboom, H.J.; Wilbrink, M.; Euverink, G.-J.W.; Dijkstra, B.W.; Dijkhuizen, L. Conversion of Cyclodextrin Glycosyltransferase into a Starch Hydrolase by Directed Evolution: The Role of Alanine 230 in Acceptor Subsite +1. Biochemistry 2003, 42, 7518–7526. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-J.; Park, C.-S.; Cho, H.-Y.; Cha, S.-S.; Kim, J.-S.; Lee, S.-B.; Moon, T.-W.; Kim, J.-W.; Oh, B.-H.; Park, K.-H. Role of the Glutamate 332 Residue in the Transglycosylation Activity of Thermus Maltogenic Amylase. Biochemistry 2000, 39, 6773–6780. [Google Scholar] [CrossRef]
- Lin, X.; Hong, T.; Mu, Y.; Torres, J. Identification of residues involved in water versus glycerol selectivity in aquaporins by differential residue pair coevolution. Biochim. Biophys. Acta Biomembr. 2012, 1818, 907–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Panchenko, A.R. Coevolution in defining the functional specificity. Proteins Struct. Funct. Bioinform. 2009, 75, 231–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vehlow, C.; Stehr, H.; Winkelmann, M.; Duarte, J.M.; Petzold, L.; Dinse, J.; Lappe, M. CMView: Interactive contact map visualization and analysis. Bioinformatics 2011, 27, 1573–1574. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, L.; De Ruvo, M.; Paci, P.; Santoni, D.; Giuliani, A. Protein Contact Networks: An Emerging Paradigm in Chemistry. Chem. Rev. 2013, 113, 1598–1613. [Google Scholar] [CrossRef]
- Roujeinikova, A.; Raasch, C.; Sedelnikova, S.; Liebl, W.; Rice, D.W. Crystallization and preliminary X-ray crystallographic studies on 4-α-glucanotransferase from Thermotoga maritima. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://swissmodel.expasy.org/assess (accessed on 14 October 2019).
- Holmes, J.B.; Tsai, J. Characterizing Conserved Structural Contacts by Pairwise Relative Contacts and Relative Packing Groups. J. Mol. Biol. 2005, 354, 706–721. [Google Scholar] [CrossRef]
- Koropatkin, N.M.; Smith, T.J. Article SusG: A Unique Cell-Membrane-Associated a -Amylase from a Prominent Human Gut Symbiont Targets Complex Starch Molecules. Struct. Des. 2010, 18, 200–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutrouli, M.; Karatzas, E.; Paez-Espino, D.; Pavlopoulos, G.A. A Guide to Conquer the Biological Network Era Using Graph Theory. Front. Bioeng. Biotechnol. 2020, 8, 34. [Google Scholar] [CrossRef]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978, 1, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Doshi, U.; Holliday, M.J.; Eisenmesser, E.Z.; Hamelberg, D. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc. Natl. Acad. Sci. USA 2016, 113, 4735–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, E.; Kaltenbach, M.; Correy, G.J.; Carr, P.D.; Porebski, B.T.; Livingstone, E.K.; Afriat-Jurnou, L.; Buckle, A.M.; Weik, M.; Hollfelder, F.; et al. The role of protein dynamics in the evolution of new enzyme function. Nat. Chem. Biol. 2016, 12, 944–950. [Google Scholar] [CrossRef]
- Van Der Kamp, M.W.; Prentice, E.J.; Kraakman, K.L.; Connolly, M.; Mulholland, A.J.; Arcus, V.L. Dynamical origins of heat capacity changes in enzyme-catalysed reactions. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Wang, L.; Su, L.; Wu, J. Effect of Leu 277 on Disproportionation and Hydrolysis Activity in Bacillus stearothermophilus NO2 Cyclodextrin Glucosyltransferase. Appl. Environ. Microbiol. 2021, 87, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins Struct. Funct. Bioinform. 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Mai, U.; Pfeiffer, W.; Janssen, S.; Asnicar, F.; Sanders, J.G.; Belda-ferre, P.; Al-ghalith, G.A.; Kopylova, E.; Mcdonald, D.; et al. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat. Commun. 2019, 10, 5477. [Google Scholar] [CrossRef] [Green Version]
- Stam, M.R.; Danchin, E.G.J.; Rancurel, C.; Coutinho, P.M.; Henrissat, B. Dividing the large glycoside hydrolase family 13 into subfamilies: Towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 2006, 19, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Janeček, S. α-amylase family: Molecular biology and evolution. Prog. Biophys. Mol. Biol. 1997, 67, 67–97. [Google Scholar] [CrossRef]
- Zhao, J.; Tandrup, T.; Bissaro, B.; Barbe, S.; Poulsen, J.N.; Andr, I.; Dumon, C.; Lo, L.; Donohue, M.J.O. Probing the determinants of the transglycosylation / hydrolysis partition in a retaining α-L-arabinofuranosidase. New Biotechnol. J. 2021, 62, 68–78. [Google Scholar] [CrossRef]
- Biswas, P.; Adhikari, A.; Pal, U.; Singh, P.; Das, M.; Saha-Dasgupta, T.; Choudhury, S.S.; Das, R.; Pal, S.K. Flexibility modulates the catalytic activity of a thermostable enzyme: Key information from optical spectroscopy and molecular dynamics simulation. Soft Matter 2020, 16, 3050–3062. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.-H.; Jung, J.-H.; Jung, D.-H.; Park, S.; Yoo, S.-H.; Kim, Y.-R.; Park, C.-S. An unusual chimeric amylosucrase generated by domain-swapping mutagenesis. Enzyme Microb. Technol. 2016, 86, 7–16. [Google Scholar] [CrossRef]
- Seo, D.-H.; Jung, J.-H.; Park, C.-S. Improved polymerization activity of Deinococcus geothermalis amylosucrase by semi-rational design: Effect of loop flexibility on the polymerization reaction. Int. J. Biol. Macromol. 2019, 130, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xiang, G.; Leemhuis, H.; van der Maarel, M.J.E.C. Structural elements determining the transglycosylating activity of glycoside hydrolase family 57 glycogen branching enzymes. Proteins Struct. Funct. Bioinform. 2021, prot.26200. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Koivula, A.K.; Lehtovaara, P.M.; Hemminki, A.; Knowles, J.K.C. Random mutagenesis used to probe the structure and function of Bacillus stearothermophilus alpha-amylase. Protein Eng. Des. Sel. 1990, 3, 181–191. [Google Scholar] [CrossRef]
- Wang, C.; Huang, R.; He, B.; Du, Q. Improving the thermostability of alpha-amylase by combinatorial coevolving-site saturation mutagenesis. BMC Bioinform. 2012, 13, 263. [Google Scholar] [CrossRef] [Green Version]
- Hleap, J.S.; Blouin, C. The response to selection in Glycoside Hydrolase Family 13 structures: A comparative quantitative genetics approach. PLoS ONE 2018, 13, e0196135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Verma, D.; Sheridan, R.P.; Liaw, A.; Ma, J.; Marshall, N.M.; McIntosh, J.; Sherer, E.C.; Svetnik, V.; Johnston, J.M. Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model. 2020, 60, 2773–2790. [Google Scholar] [CrossRef]
- Narayanan, H.; Dingfelder, F.; Butté, A.; Lorenzen, N.; Sokolov, M.; Arosio, P. Machine Learning for Biologics: Opportunities for Protein Engineering, Developability, and Formulation. Trends Pharmacol. Sci. 2021, 42, 151–165. [Google Scholar] [CrossRef]
- Timonina, D.; Sharapova, Y.; Švedas, V.; Suplatov, D. Bioinformatic analysis of subfamily-specific regions in 3D-structures of homologs to study functional diversity and conformational plasticity in protein superfamilies. Comput. Struct. Biotechnol. J. 2021, 19, 1302–1311. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Holm, L. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- James, M.; Murtola, T.; Schulz, R.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Softw. X 2015, 2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Sefidbakht, Y.; Siadat, O.R.; Taheri, F. Homology modeling and molecular dynamics study on Schwanniomyces occidentalis alpha- amylase. J. Biomol. Struct. Dyn. 2016, 35, 574–584. [Google Scholar] [CrossRef]
- Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 26 July 2021).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Kinh, R.; Do, G.; Andrej, S. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Grace. Available online: http://plasma-gate.weizmann.ac.il/Grace/ (accessed on 10 July 2021).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. ProMod3—A versatile homology modelling toolbox. PLOS Comput. Biol. 2021, 17, e1008667. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the local structural quality of transmembrane protein models using statistical potentials (QMEANBrane). Bioinformatics 2014, 30, i505–i511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [Green Version]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A Method to Identify Protein Sequences That Fold into a Known Three-Dimensional Structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins Struct. Funct. Genet. 1993, 17, 355–362. [Google Scholar] [CrossRef]
- Sarkar, G.; Sommer, S.S. The “megaprimer” method of site-directed mutagenesis. Biotechniques 1990, 8, 404–407. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Miller, G.L. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- Xiao, Z.; Storms, R.; Tsang, A. A quantitative starch-iodine method for measuring alpha-amylase and glucoamylase activities. Anal. Biochem. 2006, 351, 146–148. [Google Scholar] [CrossRef]
- Zeeman, S.C.; Tiessen, A.; Pilling, E.; Kato, K.L.; Donald, A.M.; Smith, A.M. Starch Synthesis in Arabidopsis. Granule Synthesis, Composition, and Structure. Plant Physiol. 2002, 129, 516–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mould, D.L.; Synge, R.L.M. Separations of polysaccharides related to starch by electrokinetic ultrafiltration in collodion membranes. Biochem. J. 1954, 58, 571–585. [Google Scholar] [CrossRef] [Green Version]
- Mould, D.L. Potentiometric and spectrophotometric studies of complexes of hydrolysis products of amylose with iodine and potassium iodide. Biochem. J. 1954, 58, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Wind, R.D.; Liebl, W.; Buitelaar, R.M.; Penninga, D.; Spreinat, A.; Dijkhuizen, L.; Bahl, H. Cyclodextrin formation by the thermostable alpha-amylase of Thermoanaerobacterium thermosulfurigenes EM1 and reclassification of the enzyme as a cyclodextrin glycosyltransferase. Appl. Environ. Microbiol. 1995, 61, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Haga, K.; Yamane, K. Four aromatic residues in the active center of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. 1011: Effects of replacements on substrate binding and cyclization characteristics. Biochemistry 1994, 33, 9929–9936. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, B.A.; van Alebeek, G.J.; Uitdehaag, J.C.; Dijkstra, B.W.; Dijkhuizen, L. The three transglycosylation reactions catalyzed by cyclodextrin glycosyltransferase from Bacillus circulans (strain 251) proceed via different kinetic mechanisms. Eur. J. Biochem. 2000, 267, 658–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.-S.; Park, J.-T.; Kang, H.-K.; Cha, H.; Kim, D.-S.; Kim, J.-W.; Park, K.-H. TreX from Sulfolobus solfataricus ATCC 35092 displays isoamylase and 4-alpha-glucanotransferase activities. Biosci. Biotechnol. Biochem. 2007, 71, 1348–1352. [Google Scholar] [CrossRef]
- Jørgensen, S.; Vorgias, C.E.; Antranikian, G. Cloning, Sequencing, Characterization, and Expression of an Extracellular α-Amylase from the Hyperthermophilic ArchaeonPyrococcus furiosus in Escherichia coli andBacillus subtilis. J. Biol. Chem. 1997, 272, 16335–16342. [Google Scholar] [CrossRef] [Green Version]
- Robyt, J.F.; French, D. The action pattern of porcine pancreatic alpha-amylase in relationship to the substrate binding site of the enzyme. J. Biol. Chem. 1970, 245, 3917–3927. [Google Scholar] [CrossRef]
- Kramhøft, B.; Bak-Jensen, K.S.; Mori, H.; Juge, N.; Nøhr, J.; Svensson, B. Involvement of individual subsites and secondary substrate binding sites in multiple attack on amylose by barley alpha-amylase. Biochemistry 2005, 44, 1824–1832. [Google Scholar] [CrossRef]
- Tonozuka, T.; Ohtsuka, M.; Mogi, S.; Sakai, H.; Ohta, T.; Sakano, Y. A neopullulanase-type alpha-amylase gene from Thermoactinomyces vulgaris R-47. Biosci. Biotechnol. Biochem. 1993, 57, 395–401. [Google Scholar] [CrossRef]
- Kanai, R.; Haga, K.; Akiba, T.; Yamane, K.; Harata, K. Biochemical and crystallographic analyses of maltohexaose-producing amylase from alkalophilic Bacillus sp. 707. Biochemistry 2004, 43, 14047–14056. [Google Scholar] [CrossRef]
- Brayer, G.D.; Sidhu, G.; Maurus, R.; Rydberg, E.H.; Braun, C.; Wang, Y.; Nguyen, N.T.; Overall, C.M.; Withers, S.G. Subsite mapping of the human pancreatic alpha-amylase active site through structural, kinetic, and mutagenesis techniques. Biochemistry 2000, 39, 4778–4791. [Google Scholar] [CrossRef]
- Choi, J.-H.; Lee, H.; Kim, Y.-W.; Park, J.-T.; Woo, E.-J.; Kim, M.-J.; Lee, B.-H.; Park, K.-H. Characterization of a novel debranching enzyme from Nostoc punctiforme possessing a high specificity for long branched chains. Biochem. Biophys. Res. Commun. 2009, 378, 224–229. [Google Scholar] [CrossRef]
- Tan, T.-C.; Mijts, B.N.; Swaminathan, K.; Patel, B.K.C.; Divne, C. Crystal structure of the polyextremophilic alpha-amylase AmyB from Halothermothrix orenii: Details of a productive enzyme-substrate complex and an N domain with a role in binding raw starch. J. Mol. Biol. 2008, 378, 852–870. [Google Scholar] [CrossRef]
- Shipman, J.A.; Cho, K.H.; Siegel, H.A.; Salyers, A.A. Physiological characterization of SusG, an outer membrane protein essential for starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 1999, 181, 7206–7211. [Google Scholar] [CrossRef] [Green Version]
- Nitta, Y.; Mizushima, M.; Hiromi, K.; Ono, S. Influence of molecular structures of substrates and analogues on Taka-amylase A catalyzed hydrolyses. I. Effect of chain length of linear substrates. J. Biochem. 1971, 69, 567–576. [Google Scholar]
- Nitschke, L.; Heeger, K.; Bender, H.; Schulz, G.E. Molecular cloning, nucleotide sequence and expression in Escherichia coli of the beta-cyclodextrin glycosyltransferase gene from Bacillus circulans strain no. 8. Appl. Microbiol. Biotechnol. 1990, 33, 542–546. [Google Scholar] [CrossRef]
- Li, Z.; Li, B.; Gu, Z.; Du, G.; Wu, J.; Chen, J. Extracellular expression and biochemical characterization of alpha-cyclodextrin glycosyltransferase from Paenibacillus macerans. Carbohydr. Res. 2010, 345, 886–892. [Google Scholar] [CrossRef]
- Violet, M.; Meunier, J.C. Kinetic study of the irreversible thermal denaturation of Bacillus licheniformis alpha-amylase. Biochem. J. 1989, 263, 665–670. [Google Scholar] [CrossRef]
- Nakada, T.; Kubota, M.; Sakai, S.; Tsujisaka, Y. Purification and characterization of two forms of maltotetraose-forming amylase from Pseudomonas stutzeri. Agric. Biol. Chem. 1990, 54, 737–743. [Google Scholar] [CrossRef]
- Tonozuka, T.; Mogi, S.; Shimura, Y.; Ibuka, A.; Sakai, H.; Matsuzawa, H.; Sakano, Y.; Ohta, T. Comparison of primary structures and substrate specificities of two pullulan-hydrolyzing alpha-amylases, TVA I and TVA II, from Thermoactinomyces vulgaris R-47. Biochim. Biophys. Acta 1995, 1252, 35–42. [Google Scholar] [CrossRef]
- Mijts, B.N.; Patel, B.K.C. Cloning, sequencing and expression of an alpha-amylase gene, amyA, from the thermophilic halophile Halothermothrix orenii and purification and biochemical characterization of the recombinant enzyme. Microbiology 2002, 148, 2343–2349. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Vinuesa, L.; Hernandez-Hernandez, O.; Moreno, F.J.; de Las Rivas, B.; Muñoz, R. Unravelling the diversity of glycoside hydrolase family 13 α-amylases from Lactobacillus plantarum WCFS1. Microb. Cell Fact. 2019, 18, 183. [Google Scholar] [CrossRef] [Green Version]
- Buedenbender, S.; Schulz, G.E. Structural base for enzymatic cyclodextrin hydrolysis. J. Mol. Biol. 2009, 385, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Zhou, P.; Hu, S.; Yang, S.; Yan, Q.; Jiang, Z. A novel multifunctional α-amylase from the thermophilic fungus Malbranchea cinnamomea: Biochemical characterization and three-dimensional structure. Appl. Biochem. Biotechnol. 2013, 170, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Park, J.-T.; Li, X.; Kim, S.; Lee, S.; Shim, J.-H.; Park, S.-H.; Cha, J.; Lee, B.-H.; Kim, J.-W.; et al. Overexpression and characterization of an extremely thermostable maltogenic amylase, with an optimal temperature of 100 degrees C, from the hyperthermophilic archaeon Staphylothermus marinus. N. Biotechnol. 2010, 27, 300–307. [Google Scholar] [CrossRef]
- Park, J.-T.; Song, H.-N.; Jung, T.-Y.; Lee, M.-H.; Park, S.-G.; Woo, E.-J.; Park, K.-H. A novel domain arrangement in a monomeric cyclodextrin-hydrolyzing enzyme from the hyperthermophile Pyrococcus furiosus. Biochim. Biophys. Acta 2013, 1834, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.-Y.; Kim, J.-S.; Choi, K.-H.; Cha, J.; Ha, N.-C. Structure of a novel α-amylase AmyB from Thermotoga neapolitana that produces maltose from the nonreducing end of polysaccharides. Acta Crystallogr. D. Biol. Crystallogr. 2013, 69, 442–450. [Google Scholar] [CrossRef]
- Tomazic, S.J.; Klibanov, A.M. Mechanisms of irreversible thermal inactivation of Bacillus alpha-amylases. J. Biol. Chem. 1988, 263, 3086–3091. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TmAmyA Variant | Hydrolysis (mEq Dextrose/µg Protein × 10−2) | Transglycosidation (mEq Butyl Glucoside/µg Protein × 10−4) | Transglycosidation/Hydrolysis (T/H) Ratio × 10−2 |
|---|---|---|---|
| Wild type | 2.8 ± 0.2 | 7 ± 1 | 2.5 ± 0.6 |
| K98A/D99P | 1.93 ± 0.09 | 5.4 ± 0.5 | 2.8 ± 0.4 |
| H222Q | 2.2 ± 0.3 | 7.9 ± 0.8 | 3.6 ± 0.4 |
| H222Q/K98A/D99P | 1.8 ± 0.1 | 10 ± 2 | 5.6 ± 0.4 |
| TmGTase Variant | Hydrolytic Activity (×10−5 mg Starch/µg Protein/min) | Transglycosidic Activity (×10−3 mg Starch/µg Protein/min) | Hydrolysis/Transglycosidation (H/T) Ratio (×10−2) |
|---|---|---|---|
| Wild type | 5.1 ± 0.3 | 4.0 ± 0.8 | 1.3 ± 0.3 |
| V86I | 2.9 ± 0.3 | 1.3 ± 0.2 | 2.2 ± 0.6 |
| M279N | 6.3 ± 0.8 | 1.01 ± 0.03 | 6 ± 1 |
| V86I/M279N | 4.2 ± 0.2 | ND | NA |
| T274V/M279N | 4.2 ± 0.3 | ND | NA |
| F72L/V86I/T274V | 4.4 ± 0.2 | 0.51 ± 0.07 | 9 ± 2 |
| F72L/V86I/T274V/M297N | 4.1 ± 0.5 | 0.65 ± 0.03 | 6 ± 1 |
| F72L/E77G/E226K/T274V | 7.0 ± 0.9 | 5 ± 1 | 1.4 ± 0.5 |
| F72L/T274V | 4.9 ± 0.5 | 1.7 ± 0.2 | 2.9 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arreola-Barroso, R.A.; Llopiz, A.; Olvera, L.; Saab-Rincón, G. Modulating Glycoside Hydrolase Activity between Hydrolysis and Transfer Reactions Using an Evolutionary Approach. Molecules 2021, 26, 6586. https://doi.org/10.3390/molecules26216586
Arreola-Barroso RA, Llopiz A, Olvera L, Saab-Rincón G. Modulating Glycoside Hydrolase Activity between Hydrolysis and Transfer Reactions Using an Evolutionary Approach. Molecules. 2021; 26(21):6586. https://doi.org/10.3390/molecules26216586
Chicago/Turabian StyleArreola-Barroso, Rodrigo A., Alexey Llopiz, Leticia Olvera, and Gloria Saab-Rincón. 2021. "Modulating Glycoside Hydrolase Activity between Hydrolysis and Transfer Reactions Using an Evolutionary Approach" Molecules 26, no. 21: 6586. https://doi.org/10.3390/molecules26216586
APA StyleArreola-Barroso, R. A., Llopiz, A., Olvera, L., & Saab-Rincón, G. (2021). Modulating Glycoside Hydrolase Activity between Hydrolysis and Transfer Reactions Using an Evolutionary Approach. Molecules, 26(21), 6586. https://doi.org/10.3390/molecules26216586