
In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies

 ,
,  , ,
, ,  and
and 
Abstract
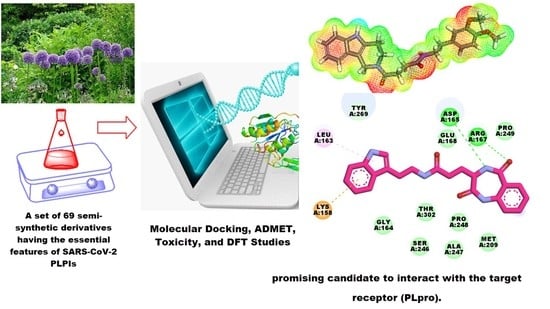
:
1. Introduction
2. Results
2.1. Docking Studies
2.2. Pharmacokinetic Profiling Study
2.3. ADMET Studies
2.4. Toxicity Studies
2.5. DFT Studies
2.5.1. Molecular Orbital Analysis
2.5.2. Molecular Electrostatic Potential Maps (MEP)
3. Conclusions
4. Method
4.1. Docking Studies
4.2. Pharmacokinetic Profiling
4.3. ADMET Analysis
4.4. Toxicity Studies
4.5. DFT Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 10 September 2021).
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Marrone, T.J.; Briggs, A.; James, M.; McCammon, J.A. Structure-based drug design: Computational advances. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 71–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Wang, Y.; Li, W.; Li, H.; Yang, L.; Wang, J.; Mahdy, H.A.; Mehany, A.; Jaiash, D.A.; Santali, E.Y. Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem. 2020, 2020, 1631243. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Fares, M.; Al-Rashood, S.T.; Al-Rashood, K.A.; Abdel-Aziz, M.M.; Soliman, D.H. Synthesis, biological evaluation and 2D-QSAR study of halophenyl bis-hydrazones as antimicrobial and antitubercular agents. Int. J. Mol. Sci. 2015, 16, 8719–8743. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.K.; Khan, F.; Negi, A.S. Pharmacophore modeling, molecular docking, QSAR, and in silico ADMET studies of gallic acid derivatives for immunomodulatory activity. J. Mol. Model. 2012, 18, 2513–2525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pei, J.; Lai, L. Computational multitarget drug design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Youssef, M.I.; Zhou, Y.; Eissa, I.H.; Wang, Y.; Zhang, J.; Jiang, L.; Hu, W.; Qi, J.; Chen, Z. Tetradecyl 2, 3-dihydroxybenzoate alleviates oligodendrocyte damage following chronic cerebral hypoperfusion through IGF-1 receptor. Neurochem. Int. 2020, 138, 104749. [Google Scholar] [CrossRef]
- Hadni, H.; Elhallaoui, M. 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J. Chem. 2020, 44, 6553–6565. [Google Scholar] [CrossRef]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg. Chem. 2021, 112, 104947. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Eissa, I.H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Elkady, H.; Elwan, A. Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1760–1782. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Alaa, E.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Taghour, M.S.; Eissa, I.H. Discovery of new 3-methylquinoxalines as potential anti-cancer agents and apoptosis inducers targeting VEGFR-2: Design, synthesis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2021, 36, 1732–1750. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Elwan, A.; Elkady, H. Discovery of new VEGFR-2 inhibitors based on bis ([1, 2, 4] triazolo)[4, 3-a: 3’, 4’-c] quinoxaline derivatives as anticancer agents and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1093–1114. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, N.A.; Dahab, M.A.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Mahdy, H.A.; Elkady, H. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: Design, molecular modeling, and synthesis. Bioorg. Chem. 2021, 110, 104807. [Google Scholar] [CrossRef]
- El-Adl, K.; Ibrahim, M.-K.; Alesawy, M.S.; Eissa, I.H. [1, 2, 4] Triazolo [4, 3-c] quinazoline and bis ([1, 2, 4] triazolo)[4, 3-a: 4′, 3′-c] quinazoline derived DNA intercalators: Design, synthesis, in silico ADMET profile, molecular docking and anti-proliferative evaluation studies. Bioorg. Med. Chem. 2021, 30, 115958. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. [Google Scholar] [CrossRef]
- Jalmakhanbetova, R.I.; Suleimen, Y.M.; Oyama, M.; Elkaeed, E.B.; Eissa, I.; Suleimen, R.N.; Metwaly, A.M.; Ishmuratova, M.Y. Isolation and In Silico Anti-COVID-19 Main Protease (Mpro) Activities of Flavonoids and a Sesquiterpene Lactone from Artemisia sublessingiana. J. Chem. 2021, 2021, 5547013. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Abdallah, A.E.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M. In Silico Studies of Some Isoflavonoids as Potential Candidates against COVID-19 Targeting Human ACE2 (hACE2) and Viral Main Protease (Mpro). Molecules 2021, 26, 2806. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; El-Aziz, A.; Mohamed, T.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- Imieje, V.O.; Zaki, A.A.; Metwaly, A.M.; Mostafa, A.E.; Elkaeed, E.B.; Falodun, A. Comprehensive In Silico Screening of the Antiviral Potentialities of a New Humulene Glucoside from Asteriscus hierochunticus against SARS-CoV-2. J. Chem. 2021, 2021, 5541876. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Ghoneim, M.M.; Eissa, I.H.; Elsehemy, I.A.; Mostafa, A.E.; Hegazy, M.M.; Afifi, W.M.; Dou, D. Traditional ancient Egyptian medicine: A review. Saudi J. Biol. Sci. 2021, 28, 5823–5832. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Y.; Metwaly, A.M.; Xue, Y.; Shi, Y.; Dou, D. The Chinese herbal formulae (Yitangkang) exerts an antidiabetic effect through the regulation of substance metabolism and energy metabolism in type 2 diabetic rats. J. Ethnopharmacol. 2019, 239, 111942. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Lianlian, Z.; Luqi, H.; Deqiang, D. Black ginseng and its saponins: Preparation, phytochemistry and pharmacological effects. Molecules 2019, 24, 1856. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-M.; Ran, X.-K.; Riaz, M.; Yu, M.; Cai, Q.; Dou, D.-Q.; Metwaly, A.M.; Kang, T.-G.; Cai, D.-C. Chemical constituents of stems and leaves of Tagetespatula L. and its fingerprint. Molecules 2019, 24, 3911. [Google Scholar] [CrossRef] [Green Version]
- Metwaly, A. Comparative biological evaluation of four endophytic fungi isolated from nigella sativa seeds. Al-Azhar J. Pharm. Sci. 2019, 59, 123–136. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Wanas, A.S.; Radwan, M.M.; Ross, S.A.; ElSohly, M.A. New α-Pyrone derivatives from the endophytic fungus Embellisia sp. Med. Chem. Res. 2017, 26, 1796–1800. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Ghoneim, M.M.; Musa, A. Two new antileishmanial diketopiperazine alkaloids from the endophytic fungus Trichosporum sp. Derpharmachemica 2015, 7, 322–327. [Google Scholar]
- Ghoneim, M.M.; Afifi, W.M.; Ibrahim, M.; Elagawany, M.; Khayat, M.T.; Aboutaleb, M.H.; Metwaly, A.M. Biological evaluation and molecular docking study of metabolites from Salvadora Persica L. Growing in Egypt. Pharmacogn. Mag. 2019, 15, 232. [Google Scholar]
- Liu, L.; Luo, S.; Yu, M.; Metwaly, A.M.; Ran, X.; Ma, C.; Dou, D.; Cai, D. Chemical Constituents of Tagetes patula and Their Neuroprotecting Action. Nat. Prod. Commun. 2020, 15, 1–8. [Google Scholar]
- Yassin, A.M.; El-Deeb, N.M.; Metwaly, A.M.; El Fawal, G.F.; Radwan, M.M.; Hafez, E.E. Induction of apoptosis in human cancer cells through extrinsic and intrinsic pathways by Balanites aegyptiaca furostanol saponins and saponin-coated silvernanoparticles. Appl. Biochem. Biotechnol. 2017, 182, 1675–1693. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, M.H.; El-Sherbiny, G.M.; Moghannem, S.A.; Abdelmonem, M.; Elsehemy, I.A.; Metwaly, A.M.; Kalaba, M.H. New combination approaches to combat methicillin-resistant Staphylococcus aureus (MRSA). Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Suleimen, Y.M.; Metwaly, A.M.; Mostafa, A.E.; Elkaeed, E.B.; Liu, H.-W.; Basnet, B.B.; Suleimen, R.N.; Ishmuratova, M.Y.; Turdybekov, K.M.; Van Hecke, K. Isolation, Crystal Structure, and In Silico Aromatase Inhibition Activity of Ergosta-5, 22-dien-3β-ol from the Fungus Gyromitra esculenta. J. Chem. 2021, 2021, 5529786. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Kadry, H.A.; Atef, A.; Mohammad, A.-E.I.; Ma, G.; Cutler, S.J.; Ross, S.A. Nigrosphaerin A a new isochromene derivative from the endophytic fungus Nigrospora sphaerica. Phytochem. Lett. 2014, 7, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metwaly, A.M.; Fronczek, F.R.; Ma, G.; Kadry, H.A.; Atef, A.; Mohammad, A.-E.I.; Cutler, S.J.; Ross, S.A. Antileukemic α-pyrone derivatives from the endophytic fungus Alternaria phragmospora. Tetrahedron Lett. 2014, 55, 3478–3481. [Google Scholar] [CrossRef] [Green Version]
- Zhanzhaxina, A.; Suleimen, Y.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Suleimen, R.; Ishmuratova, M.; Akatan, K.; Luyten, W. In Vitro and In Silico Cytotoxic and Antibacterial Activities of a Diterpene from Cousinia alata Schrenk. J. Chem. 2021, 2021, 5542455. [Google Scholar] [CrossRef]
- Imieje, V.O.; Zaki, A.A.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Ali, Z.; Khan, I.A.; Falodun, A. Antileishmanial Derivatives of Humulene from Asteriscus hierochunticus with in silico Tubulin Inhibition Potential. Rec. Nat. Prod. 2021, 16, 150–171. [Google Scholar]
- Jalmakhanbetova, R.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M.; Suleimen, Y.M. Synthesis and Molecular Docking of Some Grossgemin Amino Derivatives as Tubulin Inhibitors Targeting Colchicine Binding Site. J. Chem. 2021, 2021, 5586515. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- da Rosa, R.; Schenkel, E.P.; Bernardes, L.S.C. Semisynthetic and newly designed derivatives based on natural chemical scaffolds: Moving beyond natural products to fight Trypanosoma cruzi. Phytochem. Rev. 2020, 19, 105–122. [Google Scholar] [CrossRef]
- Sánchez-Recillas, A.; Navarrete-Vázquez, G.; Hidalgo-Figueroa, S.; Rios, M.Y.; Ibarra-Barajas, M.; Estrada-Soto, S. Semisynthesis, ex vivo evaluation, and SAR studies of coumarin derivatives as potential antiasthmatic drugs. Eur. J. Med. Chem. 2014, 77, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; John, S.E.S.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Báez-Santos, Y.M.; Barraza, S.J.; Wilson, M.W.; Agius, M.P.; Mielech, A.M.; Davis, N.M.; Baker, S.C.; Larsen, S.D.; Mesecar, A.D. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014, 57, 2393–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.E.; Alesawy, M.S.; Eissa, S.I.; El-Fakharany, E.M.; Kalaba, M.H.; Sharaf, M.H.; Shama, N.M.A.; Mahmoud, S.H.; Mostafa, A.; Al-Karmalawy, A.A.; et al. Design and synthesis of new 4-(2-nitrophenoxy) benzamide derivatives as potential antiviral agents: Molecular modeling and in vitro antiviral screening. New J. Chem. 2021, 45, 16557–16571. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; Oualid, F.E.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Laboratory, E. Natural-Product-Based Library. Available online: https://eximedlab.com/Screening-Compounds.html (accessed on 11 August 2021).
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA. QSAR, ADMET and Predictive Toxicology. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 11 September 2021).
- Venkatapathy, R.; Wang, N.C.Y.; Martin, T.M.; Harten, P.F.; Young, D. Structure–Activity Relationships for Carcinogenic Potential. Gen. Appl. Syst. Toxicol. 2009. [Google Scholar] [CrossRef]
- Goodrnan, G.; Wilson, R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar] [CrossRef]
- Council, N.R. Correlation between carcinogenic potency and the maximum tolerated dose: Implications for risk assessment. In Issues in Risk Assessment; National Academies Press (US): Washington, DC, USA, 1993. [Google Scholar]
- Gonella Diaza, R.; Manganelli, S.; Esposito, A.; Roncaglioni, A.; Manganaro, A.; Benfenati, E. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res. 2015, 26, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, F.; Benfenati, E. In silico models for repeated-dose toxicity (RDT): Prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs. In In Silico Methods for Predicting Drug Toxicity; Springer: Berlin/Heidelberg, Germany, 2016; pp. 163–176. [Google Scholar]
- Venkatapathy, R.; Moudgal, C.J.; Bruce, R.M. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J. Chem. Inf. Comput. Sci. 2004, 44, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, K.R. The Draize eye test. Surv. Ophthalmol. 2001, 45, 493–515. [Google Scholar] [CrossRef]
- Bosshard, E. Review on skin and mucous-membrane irritation tests and their application. Food Chem. Tox. 1985, 23, 149–154. [Google Scholar] [CrossRef]
- Subashchandrabose, S.; Saleem, H.; Erdogdu, Y.; Rajarajan, G.; Thanikachalam, V. FT-Raman, FT-IR spectra and total energy distribution of 3-pentyl-2, 6-diphenylpiperidin-4-one: DFT method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 82, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Aguilar, C.A.; Palos-Barba, V.; Thangarasu, P.; Koodali, R.T. Visible light driven photo-degradation of Congo red by TiO2ZnO/Ag: DFT approach on synergetic effect on band gap energy. Chemosphere 2018, 213, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.S.; Tripathi, V.D.; Darghouth, A.A. Synthesis, Characterization, DFT calculation and Antimicrobial Activity of Co (II) and Cu (II) complexes with azo dye. In Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2019; p. 052051. [Google Scholar]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; Wiley: Hoboken, NJ, USA, 1977. [Google Scholar]
- El-Nahass, M.; Kamel, M.; El-Deeb, A.; Atta, A.; Huthaily, S. Ab initio HF, DFT and experimental (FT-IR) investigation of vibrational spectroscopy of PN, N-dimethylaminobenzylidenemalononitrile (DBM). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 443–450. [Google Scholar] [CrossRef]
- Aihara, J.-I. Correlation found between the HOMO–LUMO energy separation and the chemical reactivity at the most reactive site for isolated-pentagon isomers of fullerenes. Phys. Chem. Chem. Phys. 2000, 2, 3121–3125. [Google Scholar] [CrossRef]
- Suhasini, M.; Sailatha, E.; Gunasekaran, S.; Ramkumaar, G. Vibrational and electronic investigations, thermodynamic parameters, HOMO and LUMO analysis on Lornoxicam by density functional theory. J. Mol. Struct. 2015, 1100, 116–128. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo Junior, W.F. Electrostatic Potential Energy in Protein-Drug Complexes. Curr. Med. Chem. 2021, 28, 4954–4971. [Google Scholar] [CrossRef] [PubMed]
- Matin, M.M.; Hasan, M.S.; Uzzaman, M.; Bhuiyan, M.M.H.; Kibria, S.M.; Hossain, M.E.; Roshid, M.H. Synthesis, spectroscopic characterization, molecular docking, and ADMET studies of mannopyranoside esters as antimicrobial agents. J. Mol. Struct. 2020, 1222, 128821. [Google Scholar] [CrossRef]
- X-ray Structural and Biological Evaluation of a Series of Potent and Highly Selective Inhibitors of Human Coronavirus Papain-like Proteases. 2021. Available online: https://www.rcsb.org/structure/4OW0 (accessed on 11 September 2021).
- El-Gamal, K.M.; El-Morsy, A.M.; Saad, A.M.; Eissa, I.H.; Alswah, M. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J. Mol. Struct. 2018, 1166, 15–33. [Google Scholar] [CrossRef]
- Yousef, R.; Sakr, H.; Eissa, I.; Mehany, A.; Metwaly, A.; Elhendawy, M.A.; Radwan, M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Amer, H.H.; Alotaibi, S.H.; Trawneh, A.H.; Metwaly, A.M.; Eissa, I.H. Anticancer activity, spectroscopic and molecular docking of some new synthesized sugar hydrazones, Arylidene and α-Aminophosphonate derivatives. Arab. J. Chem. 2021, 14, 103348. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Al-Karmalawy, A.A.; Elkaeed, E.B.; Alswah, M.; Belal, A.; Taghour, M.S.; Eissa, I.H. Design and discovery of new 1, 2, 4-triazolo [4, 3-c] quinazolines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Der Pharm. 2021, 354, 2000237. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Khalifa, M.M.; Elkaeed, E.B.; Hafez, E.E.; Alsfouk, A.A.; Metwaly, A.M. In Silico Exploration of Potential Natural Inhibitors against SARS-CoV-2 nsp10. Molecules 2021, 26, 6151. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | ΔG | Comp. | ΔG |
|---|---|---|---|
| 1 | −7.55 | 36 | −7.49 |
| 2 | −7.66 | 37 | −7.47 |
| 3 | −8.07 | 38 | −8.04 |
| 4 | −7.27 | 39 | −7.92 |
| 5 | −7.54 | 40 | −8.54 |
| 6 | −7.78 | 41 | −8.39 |
| 7 | −6.72 | 42 | −8.22 |
| 8 | −7.44 | 43 | −8.53 |
| 9 | −8.06 | 44 | −7.79 |
| 10 | −6.79 | 45 | −7.47 |
| 11 | −7.42 | 46 | −7.67 |
| 12 | −6.73 | 47 | −8.57 |
| 13 | −7.57 | 48 | −7.22 |
| 14 | −7.35 | 49 | −7.28 |
| 15 | −7.54 | 50 | −8.13 |
| 16 | −8.08 | 51 | −8.27 |
| 17 | −8.50 | 52 | −7.59 |
| 18 | −8.05 | 53 | −7.63 |
| 19 | −7.34 | 54 | −8.33 |
| 20 | −6.55 | 55 | −7.60 |
| 21 | −7.08 | 56 | −7.10 |
| 22 | −7.19 | 57 | −8.34 |
| 23 | −5.80 | 58 | −8.65 |
| 24 | −6.15 | 59 | −8.01 |
| 25 | −7.98 | 60 | −8.22 |
| 26 | −6.86 | 61 | −7.58 |
| 27 | −6.05 | 62 | −7.98 |
| 28 | −8.48 | 63 | −7.50 |
| 29 | −8.12 | 64 | −8.07 |
| 30 | −7.48 | 65 | −8.33 |
| 31 | −8.33 | 66 | −8.15 |
| 32 | −8.11 | 67 | −7.86 |
| 33 | −7.50 | 68 | −8.20 |
| 34 | −8.97 | 69 | −8.18 |
| 35 | −7.72 | ligandS88 | −8.59 |
| Compound | Lipinski’s Rule of 5 | Veber’s Rule | |||||
|---|---|---|---|---|---|---|---|
| Log P | Mole. Wt. | HBD | HBA | Violation | Number of Rotatable Bonds | TPSA | |
| of Lipinski’s Rule | |||||||
| 17 | 3.38 | 441.47 | 1 | 7 | 0 | 9 | 92.32 |
| 28 | 2.83 | 435.51 | 2 | 4 | 0 | 8 | 83.66 |
| 31 | 5.69 | 485.5 | 2 | 4 | 1 | 7 | 88.77 |
| 34 | 1.8 | 390.43 | 4 | 3 | 0 | 6 | 103.09 |
| 40 | 1.46 | 389.46 | 1 | 5 | 0 | 7 | 109 |
| 41 | 1.96 | 426.46 | 1 | 6 | 0 | 7 | 86.33 |
| 43 | 1.73 | 449.5 | 1 | 6 | 0 | 9 | 98.58 |
| 47 | 3 | 380.43 | 1 | 4 | 0 | 8 | 69.56 |
| 54 | 2.7 | 390.47 | 2 | 2 | 0 | 5 | 68.44 |
| 58 | 1.56 | 472.6 | 3 | 2 | 0 | 6 | 72.88 |
| 65 | 3.41 | 465.97 | 2 | 3 | 0 | 4 | 71.68 |
| S88 | 3.09 | 391.5 | 2 | 1 | 0 | 5 | 33.54 |
| Comp. No. | BBB Level 1 | Absorption Level 2 | Solubility | CYP2D6 4 | Hepatotoxicity Probability 5 | PPB 6 |
|---|---|---|---|---|---|---|
| Level 3 | ||||||
| 17 | ++ | +++ | ++ | −ve | 0.549 | 2 |
| 28 | ++ | +++ | +++ | −ve | 0.37 | 0 |
| 31 | + | ++ | + | +ve | 0.629 | 2 |
| 34 | ++ | +++ | +++ | +ve | 0.47 | 1 |
| 40 | ++ | +++ | +++ | −ve | 0.437 | 0 |
| 41 | ++ | +++ | +++ | −ve | 0.821 | 2 |
| 43 | ++ | +++ | +++ | −ve | 0.622 | 0 |
| 47 | +++ | +++ | ++ | +ve | 0.456 | 2 |
| 54 | +++ | +++ | +++ | −ve | 0.092 | 0 |
| 58 | ++ | +++ | +++ | −ve | 0.324 | 2 |
| 65 | +++ | +++ | ++ | −ve | 0.271 | 2 |
| S88 | ++++ | +++ | ++ | +ve | 0.092 | 1 |
| Favipiravir | ++ | +++ | ++++ | −ve | 0.728 | 0 |
| Comp. | FDA Rat Carcinogenicity 1 | Carcinogenic Potency TD50 (Rat) 2 | Rat Maximum Tolerated Dose (Feed) 3 | Rat Oral LD50 3 | Rat Chronic LOAEL 3 | Ocular Irritancy | Skin Irritancy |
|---|---|---|---|---|---|---|---|
| 17 | −ve | 5.577 | 0.142 | 4.097 | 0.039 | Mild | None |
| 28 | −ve | 1.199 | 0.144 | 27.190 | 0.539 | Mild | None |
| 31 | −ve | 5.323 | 0.369 | 0.251 | 0.043 | Mild | None |
| 34 | −ve | 0.826 | 0.328 | 32.393 | 0.232 | Mild | None |
| 40 | −ve | 10.310 | 0.033 | 14.820 | 0.339 | Mild | None |
| 41 | −ve | 1.162 | 0.122 | 26.756 | 0.446 | Mild | None |
| 43 | −ve | 1.860 | 0.100 | 26.741 | 0.151 | Mild | None |
| 47 | −ve | 14.544 | 0.045 | 8.150 | 0.139 | Mild | None |
| 54 | +ve | 0.324 | 0.068 | 5.277 | 0.015 | Mild | None |
| 58 | +ve | 1.725 | 0.071 | 5.145 | 0.001 | Mild | Mild |
| 65 | −ve | 0.315 | 0.117 | 2.971 | 0.018 | Mild | None |
| S88 | +ve | 2.548 | 0.124 | 1.229 | 0.035 | Mild | None |
| Name | Total Energy (kcal/mol) | Binding Energy (kcal/mol) | HOMO Energy (kcal/mol) | LUMO Energy (kcal/mol) | Dipole Mag | Band Gap Energy (kcal/mol) |
|---|---|---|---|---|---|---|
| 28 | −1422.912 | −12.075 | −0.170 | −0.036 | 2.790 | 0.134 |
| 34 | −1285.184 | −10.458 | −0.175 | −0.076 | 1.558 | 0.099 |
| 47 | −1252.334 | −10.395 | −0.172 | −0.075 | 2.249 | 0.097 |
| S88 | −1242.947 | −11.176 | −0.292 | −0.187 | 3.542 | 0.105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alesawy, M.S.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I.H. In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules 2021, 26, 6593. https://doi.org/10.3390/molecules26216593
Alesawy MS, Elkaeed EB, Alsfouk AA, Metwaly AM, Eissa IH. In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules. 2021; 26(21):6593. https://doi.org/10.3390/molecules26216593
Chicago/Turabian StyleAlesawy, Mohamed S., Eslam B. Elkaeed, Aisha A. Alsfouk, Ahmed M. Metwaly, and Ibrahim H. Eissa. 2021. "In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies" Molecules 26, no. 21: 6593. https://doi.org/10.3390/molecules26216593
APA StyleAlesawy, M. S., Elkaeed, E. B., Alsfouk, A. A., Metwaly, A. M., & Eissa, I. H. (2021). In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules, 26(21), 6593. https://doi.org/10.3390/molecules26216593