
Biomedical Applications of Translational Optical Imaging: From Molecules to Humans

Abstract
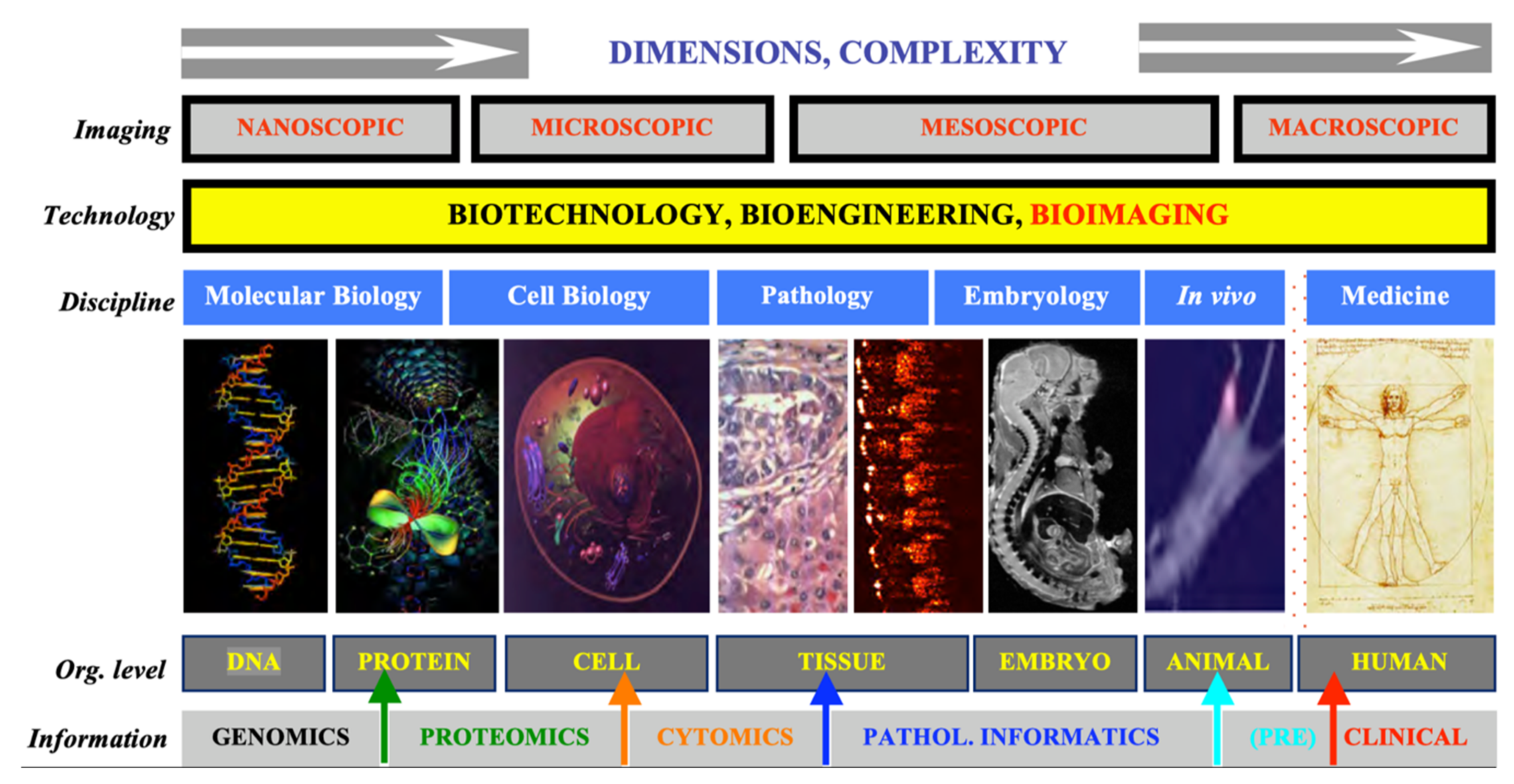
:1. Introduction
“It would be madness and inconsistency to suppose that things not yet done can be done except by means not yet tried.”Francis Bacon
2. Optical Microscopy and Its Applications
“To develop drugs for people, we basically dismantle the system. In the lab, we look at things the size of a cell or two. We dismantle life into very small models.”Aaron Ciechanover (Nobel laureate)
2.1. Confocal Microscopy
2.1.1. Membrane Electrical Potentials
2.1.2. Topologically Resolved Epigenetics by 3D Quantitative DNA Methylation Imaging
2.2. Standing Wave Microscopy: Towards Omnidirectional Superresolution
- (1)
- In the first class are methods (STED, SAX, SPEM, RESOLFT, PALM, and others) that manipulate the signal generation within the sample. For example, in photoactivated localization microscopy (PALM), the density of fluorophores is controlled so that one light emission point is activated at a time within the volume covered by the point spread function (PSF) of the imaging system. Under these conditions, the location of the fluorophore can be determined to a very high precision, which is only limited by the signal-to-noise ratio of the camera. Repeatedly activating different sets of fluorophores allows the assembly of a high-resolution image from the individual point source location maps. Other methods in this class use the saturation of fluorophores, non-linear quantum-effects such as the stimulated emission depletion (STED) via a second illumination wavelength, or the blinking of emitters (quantum dots). All these methods require extreme stability of the microscope, long acquisition times, specialized protocols and/or fluorophores, and/or high-power illumination sources that can cause severe photobleaching. These have been reviewed extensively [27,28,29,30,31], and have been constantly improved and supplemented, yielding impressive performance (see e.g., [32]).
- (2)
- In the second class, methods try to subdivide the PSF by using specialized illumination systems that use interference effects to narrow the point spread function of the microscope. As will become apparent below, structured illumination systems effectively increase the numerical aperture to improve resolution. One approach to increase the numerical aperture is to use two objectives to observe the sample from both sides simultaneously. If both images are combined optically and in a coherent fashion, the effective numerical aperture is doubled, which leads to 4π microscopy. Unfortunately, this is achieved with extreme alignment difficulties, which makes 4π microscopy rather impractical and expensive (see [27,30,31]).
2.3. Multimode Microscopy
2.3.1. Live Cell Motion
2.3.2. Cancer Stem Cells
2.4. Hyperspectral Microscopy for Clinical Diagnostics
2.4.1. Cytopathology
2.4.2. Histopathology/Immunohistochemistry/Immunofluorescence
2.4.3. Cell-Level Drug Candidate Analysis—Towards High Content Screening
2.4.4. Intracellular Proteomics
2.5. Multispectral Multimode Microscopy
3. Pre-Clinical Optical Bioimaging
“But the inadequacy of these microscopes, for the observation of any but the most minute bodies, and even those if part of a larger body, destroys their utility; for if the invention could be extended to greater bodies, or the minute part of greater bodies, so that...the latent minutiae and irregularities of liquids, urine, blood, wounds, and many other things could be rendered visible, the greatest advantage would, without doubt, be derived.”Francis Bacon [5].
3.1. In Vivo Fluorescence Imaging of Cancer
Fluorochrome-Labeled Antibody Targeting for Molecular Imaging In Vivo

3.2. Multimode Imaging In Vivo
3.3. Intrinsic Imaging without Contrast Agents
3.4. Coherence-Based Imaging
4. Functional Imaging
“Science is driven by ideas, but paradigm shifts are often a direct result of advances in technology.”Peter C. Doherty (Nobel laureate)
4.1. Intrasurgical Topological Guidance: Hirschsprung’s Disease
4.2. Imaging Stem Cells In Vivo

4.3. Neuroimaging Primary Events: Calcium Transients at the Neuromuscular Junction
4.4. Oxygenation Mapping
5. Clinical Photonic Imaging
“Progress in science depends on new techniques, new discoveries and new ideas, probably in that order.”Sydney Brenner, Nobel laureate
5.1. Neurodegeneration Imaging
- You cannot effectively fight what you do not understand.
- You cannot properly understand what you cannot visualize.
- Best way to visualize is by imaging, with high spatio-temporal discrimination
- Thus, the recipe is that we need to image
- The right thing (primary pathology/biomarkers)
- In the right organism (humans, not mice)
- At the right time (as early as possible in the disease evolution)
- In the right place (best area to look, even if not prevalent, e.g., retina for AD)
- The right way (i) non-invasively; (ii) with high resolution (spatial, temporal); (iii) dynamically/repetitively; (iv) intrinsically (without added contrast agent); (v) sensitively; (vi) vs. other biomarkers (e.g., vasculature, in the case of AD), imaged simultaneously; (vii) with depth penetration.
- –
- more than half of all academic research focuses on non-primary pathologies (see 4a above)
- –
- most of academic research is in animal models (4b)
- –
- all clinical research is in late-stage disease (4c)
- –
- most of both academic and clinical research is in the brain (4d)
- –
- therefore, spatio-temporal resolution is poor (4e.ii)
- –
- all imaging is contrast agent-based (4e.iv) (but still better than subjective evaluations)
- –
- amyloid plaques (primary pathology) (4a), using curcumin (biomarker) fluorescence
- –
- in both mice and humans, translationally (4b)
- –
- early in the disease (in mice) (4c)
- –
- in the retina, in both animals and humans (4d)
- –
- non-invasively, with high resolution, dynamically, and repetitively (4e.i–iii)
- –
- imaging more of the retina (by larger angle scanning) (4d)
- –
- imaging without contrast agent (intrinsic signal: autofluorescence) (4e.iv)
- –
- imaging with higher sensitivity (for earlier detection) (4e.v)
- –
- imaging hyperspectrally (for better discrimination, and vs. e.g., vasculature) (4e.vi)
- –
- A new, laser-based hyperspectral excitation, allowing unique image acquisition
- –
- A new [161], quad-galvo technology in CSLO scanning, allowing flexible positioning of the beam’s pivot point at the eye’s pupil, providing a wider field of view than existing systems.
- –
- A new [162] photon detection technology, based on an unusual amplification scheme
- –
- 3D imaging capability over the entire thickness of the retina with cellular resolution is provided. Confocal imaging provides high spatial resolution, and because complicated scan lenses are not required in the quad-galvo design, the optical system is relatively simple but has higher NA.
- –
- Simultaneous (multimode) measurement of oxygenation, microvasculature structure, and plaque autofluorescence with our MM-CSLO should allow assessing treatment-induced changes in these and thus provide new insight into AD progression and even etiology.
- –
- Investigation of the chronological order of neurovascular disfunction and AD. For example, it is still not known whether there is a correlation between vascular damage/oxygenation changes and AD, as some hypothesized. We believe we can address this in new ways.
5.2. Dermatological Imaging
5.3. Endoscopic Imaging
5.3.1. Hyperspectral Imaging of Mucosal Surfaces in Patients
5.3.2. Elastic Scattering Imaging Endoscopy
- –
- Able to acquire the entire spectral range from 350–800nm (at 10 nm increments), in less than 0.5 s (8 ms per band). Spectral bands do not need to be in any particular order or be spaced a certain way, and indeed bands of interest can be repeated at different points during the scan.
- –
- Built for non-contact operation. The tip optics have a focal length of 10 mm, and a viewing area of about 8 mm in diameter at that range. No contact with the target is required or even desired. Indeed, the ability to image without direct specimen contact is especially useful since some tissues are easily traumatized.
- –
- Able to acquire of both parallel and perpendicular images simultaneously, allowing for these contrast mechanisms to be explored
- –
- Able to be used as a daughter endoscope in the instrument channel of an existing endoscope, to view lung or GI tissues in vivo
- –
- Able to be sterilized appropriately for use in a hospital setting
- –
- Using fast band-sequential spectral acquisition that limits intra-band interference and noise, as well as limiting movement artifacts; exposure times can be set so that the spectral image datacube has homogeneous signal-to-noise.
- –
- Able to be transported to the site of imaging/OR.
5.3.3. Multimode Imaging System for Minimally Invasive Surgery
5.4. Theranostics
5.4.1. Intraoperative Neurophotonic Detection and Guidance
5.4.2. Surgical Theranostics: Coupling Detection and Intervention in Time and Space
5.5. Sensitivity and Specificity
5.6. The Operating Room of the Future
6. Discussion
“The future is here. It is just not evenly distributed.”William Gibson
6.1. Optical Bioimaging Adoption in Medicine and Surgery
6.2. Translational Biophotonics: Moving into the Operating Room
6.3. Avoiding Errors
6.4. Biophotonics in Pandemic Times
7. Conclusions
“The best way to predict the future is to create it.”Dennis Gabor (Nobel laureate)
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujimoto, J.; Farkas, D.L. (Eds.) Biomedical Optical Imaging; Oxford University Press: Oxford, UK, 2009; 422p. [Google Scholar]
- Johnson, J.P.; King, W.A.; Farkas, D.L. Computerized imaging-guided endoscopic neurosurgery. Glob. Surg. 2006, 56–58. [Google Scholar]
- Farkas, D.L.; Demetriou, A.A. New surgery for better outcomes: Factors shaping it, and the need for high technology. Glob. Surg. 2003, 21–25. [Google Scholar]
- Farkas, D.L. Invention and commercialization in Optical Bioimaging. Nat. Biotechnol. 2009, 21, 1269–1271. [Google Scholar] [CrossRef]
- Bacon, F. Novum Organum; Liberty Fund, Inc.: Indianapolis, IN, USA, 1620; p. 102. Available online: https://oll-resources.s3.us-east-2.amazonaws.com/oll3/store/titles/1432/Bacon_0415_EBk_v6.0.pdf (accessed on 2 April 2021).
- Minsky, M. Microscopy Apparatus. U.S. Patent 3,013,467, 19 December 1961. [Google Scholar]
- Pawley, J. (Ed.) Handbook of Biological Confocal Microscopy, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2006; 1013p. [Google Scholar]
- Paddock, S. (Ed.) Confocal Microscopy: Methods and Protocols, 2nd ed.; Humana Press: Totowa, NJ, USA, 2013; 392p. [Google Scholar]
- Montana, V.; Farkas, D.L.; Loew, L.M. Dual-wavelength ratiometric fluorescence measurement of membrane potential. Biochemistry 1989, 28, 4536–4539. [Google Scholar] [CrossRef]
- Focht, D.; Fakas, D.L. Mammalian live-cell microscopy environmental control. Cell Vis. 1995, 2, 450–454. [Google Scholar]
- Farkas, D.L.; Wei, M.-D.; Febbroriello, P.; Carson, J.H.; Loew, L.M. Simultaneous imaging of cell and mitochondrial membrane potential. Biophys. J. 1989, 56, 1053–1069. [Google Scholar] [CrossRef] [Green Version]
- Ehrenberg, B.; Montana, V.; Wei, M.-D.; Wuskell, J.P.; Loew, L.M. Membrane potential can be determined in individual cells from the Nernstian distribution of cationic dyes. Biophys. J. 1988, 53, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Fink, C.; Morgan, F.; Loew, L.M. Intracellular fluorescent probe concentrations by confocal microscopy. Biophys. J. 1998, 75, 1648–1658. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.M.; Segawa, M.; Shirihai, O.S.; Liesa, M. Method for live cell super-resolution imaging of mitochondrial cristae and quantification of submitochondrial membrane potentials. Methods Cell Biol. 2020, 155, 545–555. [Google Scholar] [PubMed]
- Rovini, A.; Heslop, K.; Morris, M.E.; Gooz, M.; Gerencser, A.A.; Maldonado, E.N. Quantitative analysis of mitochondrial membrane potential heterogeneity inn unsynchronized and synchronized cancer cells. FASEB J. 2021, 35, e21148. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Du, H. Mitochondrial permeability transition: A pore intertwines brain aging and Alzheimer’s Disease. Cells 2021, 10, 649. [Google Scholar] [CrossRef]
- Tajbakhsh, J.; Wawrowsky, K.A.; Gertych, A.; Bar-Nur, O.; Vishnevsky, E.; Lindsley, E.H.; Farkas, D.L. Characterization of tumor cells and stem cells by differential nuclear methylation imaging. Prog. Biomed. Opt. Imaging 2008, 68590F, 1–10. [Google Scholar]
- Gertych, A.; Farkas, D.L.; Vishnewsky, E.; Lindsley, E.; Wawrowsky, K.A.; Tajbakhsh, J. Automated assessment of homogeneity within a cell population using 3-D mapping of nuclear signatures and Kullback-Leibler’s divergence. Cytom. A 2009, 75A, 569–583. [Google Scholar] [CrossRef] [Green Version]
- Gertych, A.; Farkas, D.L.; Tajbakhsh, J. Measuring topology of low-intensity DNA methylation sites for high throughput assessment of epigenetic drug-induced effects in cancer cells. Exp. Cell Res. 2010, 316, 3150–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajbakhsh, J.; Gertych, A.; Farkas, D.L. Utilising 3D nuclear DNA methylation patterns in cell-based assays for epigenetic drug screening. Drug Discov. World 2010, 27–35. [Google Scholar]
- Oh, J.-H. Investigation of Epigenetics in Cell Populations Using Three-Dimensional Quantitative DNA Methylation Imaging and Analysis. Ph.D. Thesis, University of Southern California, Los Angeles, CA, USA, 2011. [Google Scholar]
- Feinberg, A. Genome-scale approaches to the epigenetics of common human disease. Virchows Arch. 2010, 456, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Szyf, M. DNA Methylation and Cancer Therapy; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Oh, J.H.; Gertych, A.; Tajbakhsh, J. Nuclear DNA methylation and chromatin condensation phenotypes are distinct between normally proliferating/aging, rapidly growing/immortal, and senescent cells. Oncotarget 2013, 4, 474–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovski, D.; Tang, G.; Wawrowsky, K.; Boston, R.C.; Lambrecht, N.; Tajbakhsh, J. Prostate cancer diagnosis using epigenetic biomarkers, 3D high-content imaging and probabilistic cell-by-cell classifiers. Oncotarget 2017, 8, 57278–57301. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, J. Covisualization of Global DNA Methylation/Hydroxymethylation and Protein Biomarkers for Ultrahigh-Definition Epigenetic Phenotyping of Stem Cells. Methods Mol Biol. 2020, 2150, 79–92. [Google Scholar] [PubMed]
- Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; Betzig, E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 2008, 5, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Shim, S.-H.; He, J.; Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 2011, 8, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Leung, B.O.; Chou, K.C. Review of super-resolution fluorescence microscopy in biology. Appl. Spectrosc. 2011, 65, 967–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sydor, A.M.; Czymmec, K.J.; Puchner, E.M.; Mennella, V. Super-resolution microscopy: From single molecules to supramolecular assemblies. Trends Cell Biol. 2015, 25, 730–748. [Google Scholar] [CrossRef] [Green Version]
- Gwosh, K.C.; Pape, J.K.; Balzarotti, F.; Hoess, P.; Ellenberg, J.; Ries, J.; Hell, S.W. MINIFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat. Methods 2020, 17, 217–224. [Google Scholar] [CrossRef]
- Bailey, B.; Farkas, D.L.; Taylor, D.L.; Lanni, F. Enhancement of axial resolution in fluorescence microscopy by standing wave excitation. Nature 1993, 366, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Lanni, F.; Bailey, B.; Farkas, D.L.; Taylor, D.L. Excitation field synthesis as a means for obtaining enhanced axial resolution in fluorescence microscopes. BioImaging 1993, 1, 187–196. [Google Scholar] [CrossRef]
- Farkas, D.L.; Bailey, B.; Lanni, F.; Taylor, D.L. New waves in light microscopy. Proc. SPIE 1994, 2137, 2–16. [Google Scholar]
- Bailey, B.; Krishnamurthi, V.; Farkas, D.L.; Taylor, D.L.; Lanni, F. 3-D imaging of biological specimens with standing wave fluorescence microscopy. Proc. SPIE 1994, 2184, 208–213. [Google Scholar]
- Nowatzyk, A.G. Structured Standing Wave Microscope. U.S. Patent 10,018,818, 10 July 2018. [Google Scholar]
- Farkas, D.L.; Hell, S. (Eds.) Frontiers in Microscopy. J. Biomed. Opt. 2001, 6, 266–331. [Google Scholar]
- Farkas, D.L.; Baxter, G.; DeBiasio, R.; Gough, A.; Nederlof, M.A.; Pane, D.; Pane, J.; Patek, D.R.; Ryan, K.W.; Taylor, D.L. Multimode light microscopy and the dynamics of molecules, cells and tissues. Annu. Rev. Physiol. 1993, 55, 785–817. [Google Scholar] [CrossRef] [PubMed]
- Farkas, D.L.; Gough, A.H.; Lanni, F.; Taylor, D.L.; OPIA. Selecting and using an electronic camera for light microscopy. Am. Lab. 1995, 25–40. [Google Scholar]
- Galbraith, W.; Farkas, D.L. Remapping disparate images for coincidence. J. Microsc. 1993, 172, 163–176. [Google Scholar] [CrossRef]
- Taylor, D.L.; Deerfield, D.W., II; Fahlman, S.E.; Gough, A.H.; Lanni, F.; Farkas, D.L. Automated Interactive Microscopy: High performance computing and communications requirements for investigating the functional dynamics of living cells. In Proceedings of the Workshop on Real-Time Application High-Performance Computational Biology Imaging, Conference Report; UIUC-BI-94-02.cr; Beckman Institute: Urbana, IL, USA; 1994. [Google Scholar]
- Taylor, D.L.; Harris, L.D.; DeBiasio, R.; Fahlman, S.E.; Farkas, D.L.; Lanni, F.; Nederlof, M.; Gough, A.H. Automated Interactive Microscopy: Measuring and manipulating the chemical and molecular dynamics of cells and tissues. Proc. SPIE 1996, 2678, 15–27. [Google Scholar]
- Burton, K.; Farkas, D.L. Telemicroscopy—Net Progress. Nature 1998, 391, 540–541. [Google Scholar] [CrossRef]
- Taylor, D.L.; DeBiasio, R.; LaRocca, G.; Pane, D.; Post, P.; Kolega, J.; Giuliano, K.; Burton, K.; Gough, A.; Dow, A.; et al. Potential of machine-vision light microscopy in toxicologic pathology. Toxicol. Pathol. 1994, 22, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.L.; Burton, K.; DeBiasio, R.L.; Giuliano, K.A.; Gough, A.H.; Leonardo, T.; Pollock, J.A.; Farkas, D.L. Automated light microscopy for the study of the brain: Cellular and molecular dynamics, development and tumorigenesis. Ann. N. Y. Acad. Sci. 1997, 820, 208–228. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.D.; Deerfield, D.W.; Fahlman, S.E.; Gough, A.H.; Lanni, F.; Farkas, D.L.; Taylor, D.L. The Automated Interactive Microscope (AIM) for investigating the functional dynamics of living cells: Status and vision. In Proceedings of the 52nd Annual Meeting of the Microscopy Society of America, San Francisco, CA, USA, 31 July–5 August 1994; pp. 160–161. [Google Scholar]
- Farkas, D.L.; Wachman, E.S.; Harris, L.D.; Gough, A.H.; Taylor, D.L. Digital microscope imaging of cell dynamics. Emerg. Appl. Fluoresc. Technol. Biophys. Cell. Imaging. CLEO 1995, 15, 274–275. [Google Scholar]
- Zhao, T.; Xiong, Y.; Chung, A.; Wachman, E.S.; Farkas, D.L. Improved 3-D cellular imaging by multispectral focus assessment. Prog. Biomed. Opt. Imaging 2005, 6, 91–101. [Google Scholar]
- Taylor, D.L. A personal perspective on High Content Screening (HCS): From the beginning. J. Biomol. Screen. 2010, 15, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBiasio, R.L.; LaRocca, G.M.; Post, P.L.; Taylor, D.L. Myosin II transport, organization and phosphorylation: Evidence for cortical flow/solation-contraction coupling during cytokinesis and cell locomotion. Mol. Biol. Cell 1996, 7, 1259–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, K.; Taylor, D.L. Traction forces of cleavage and locomotion during cytokinesis on optically modified elastic substrata. Nature 1997, 385, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Blum, C.; Cesa, Y.; Escalante, M.; Subramanian, V. Multimode microscopy: Spectral and lifetime imaging. J. R. Soc. Interface 2009, 6, S35–S43. [Google Scholar] [CrossRef] [Green Version]
- Surawicz, T.S.; Davis, F.; Freels, S.; Laws, E.R., Jr.; Menck, H.R. Brain tumor survival: Results from the National Cancer Data Base. J. Neurooncol. 1998, 40, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Yuan, X.; Belladonna, M.L.; Ong, J.M.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Induction of potent antitumor immunity by intratumoral injection of interleukin 23-transduced dendritic cells. Cancer Res. 2006, 66, 8887–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Yuan, X.; Tunici, P.; Liu, G.; Xu, M.; Hu, J.; Hwang, J.Y.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of tumor stem-like cells from benign tumors. Br. J. Cancer 2009, 21, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef]
- Levenson, R.M.; Farkas, D.L. Digital spectral imaging for histopathology and cytopathology. Proc. SPIE 1997, 2983, 123–135. [Google Scholar]
- Levenson, R.M.; Wachman, E.S.; Niu, W.; Farkas, D.L. Spectral imaging in biomedicine: A selective overview. Proc. SPIE 1999, 3438, 249–259. [Google Scholar]
- Levenson, R.M.; Balestreire, E.M.; Farkas, D.L. Spectral imaging: Prospects for pathology. In Applications of Optical Engineering to the Study of Cellular Pathology; Kohen, E., Ed.; Research Signpost: Trivandrum, India, 1999; pp. 133–149. [Google Scholar]
- Farkas, D.L.; Du, C.; Fisher, G.W.; Lau, C.; Niu, W.; Wachman, E.S.; Levenson, R.M. Non-invasive image acquisition and advanced processing in optical bioimaging. Computer. Med. Imaging Graph. 1998, 22, 89–102. [Google Scholar] [CrossRef]
- Farkas, D.L. Spectral microscopy for quantitative cell and tissue imaging. In Methods in Cellular Imaging; Periasamy, A., Ed.; Oxford University Press: Oxford, UK, 2001; pp. 345–361. [Google Scholar]
- Burton, K.; Jeong, J.; Wachsmann-Hogiu, S.; Farkas, D.L. Spectral optical imaging in biology and medicine. In Biomedical Optical Imaging; Fujimoto, J., Farkas, D.L., Eds.; Oxford University Press: Oxford, UK, 2009; pp. 29–72. [Google Scholar]
- Zhao, T.; Wachman, E.S.; Farkas, D.L. A novel scheme for abnormal cell detection in Pap smear images. Prog. Biomed. Opt. Imaging 2004, 5, 151–162. [Google Scholar]
- Zhao, T.; Wachman, E.S.; Geyer, S.J.; Farkas, D.L. A recursive spectral selection scheme for unsupervised segmentation of multispectral Pap smear image sets. Prog. Biomed. Opt. Imaging 2004, 5, 175–186. [Google Scholar]
- Taube, G. Spectral technique paints cells in vivid new colors. Science 1997, 276, 1990. [Google Scholar] [CrossRef] [PubMed]
- Farkas, D.L.; Wachman, E.S.; Wachman, J.; Farkas, M.; Geyer, S. Automated Pap Screening Using Fluorescence with HPV and Multispectral Imaging. U.S. Patent 7,316,904, 8 January 2008. [Google Scholar]
- Wachman, E.S.; Niu, W.; Farkas, D.L. Imaging acousto-optic tunable filter with 0.35-micrometer spatial resolution. Appl. Opt. 1996, 35, 5220–5226. [Google Scholar] [CrossRef]
- Wachman, E.S.; Farkas, D.L.; Niu, W. Submicron Imaging System Having an Acousto-Optic Tunable Filter. U.S. Patent 5,796,512, 18 August 1998. [Google Scholar]
- Wachman, E.S.; Farkas, D.L.; Niu, W. Light Microscope Having Acousto-Optic Tunable Filters. U.S. Patent 5,841,577, 24 November 1998. [Google Scholar]
- Panell, C.N.; Wachman, E.S.; Farkas, D.L.; Ward, J.; Seale, W. Acousto-optic tuneable filters: Advances and applications to microscopy. Prog. Biomed. Opt. Imaging 2006, 7, 60880Y. [Google Scholar]
- Wachman, E.S.; Niu, W.; Farkas, D.L. AOTF microscope for imaging with increased speed and spectral versatility. Biophys. J. 1997, 73, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Plymale, D.R.; Haskins, J.R.; de la Iglesia, F.A. Monitoring simultaneous subcellular events in vitro by means of coherent multiprobe fluorescence. Nat. Med. 1999, 5, 351–355. [Google Scholar] [CrossRef]
- De la Iglesia, F.; Haskins, J.; Farkas, D.; Bearman, G. Coherent multiprobes and quantitative spectroscopic multimode microscopy for the study of simultaneous intracellular. Cytometry 2000, 39 (Suppl. S10), 34–35. [Google Scholar]
- Pollice, A.A.; Smith, C.A.; Brown, K.; Farkas, D.L.; Silverman, J.F.; Shackney, S.E. Multiparameter analysis of human epithelial tumors by laser scanning cytometry. Commun. Clin. Cytom. 2000, 42, 347–356. [Google Scholar] [CrossRef]
- Smith, C.A.; Pollice, A.; Emlet, D.; Shackney, S.E.; Pollice, A.A.; Brown, K.; Farkas, D.L.; Silverman, J.F. A simple correction for cell autofluorescence for multiparameter cell-based analysis of human solid tumors. Multiparameter analysis of human epithelial tumor cell lines by laser scanning cytometry. Cytom. B Clin. Cytom. 2006, 70, 91–103. [Google Scholar] [CrossRef]
- Wachsmann-Hogiu, S.; Annala, A.J.; Farkas, D.L. Laser applications in biology and biotechnology. In Handbook of Laser Technology and Applications; Webb, C., Ed.; Institute of Physics Publishing: Bristol, UK, 2003; pp. 2123–2155. [Google Scholar]
- Wachsmann-Hogiu, S.; Farkas, D.L. Spectroscopic non-linear microscopy. In Handbook of Biomedical Nonlinear Microscopy; So, P., Masters, B., Eds.; Oxford University Press: New York, NY, USA, 2008; pp. 461–483. [Google Scholar]
- Rativa, D.J.; Gomes, A.S.L.; Wachsmann-Hogiu, S.; Farkas, D.L.; de Araújo, R.E. Silver nanoparticles in nonlinear microscopy. In Proceedings of the Microwave and Optoelectronics Conference, Salvador, Brazil, 29 October–1 November 2007; pp. 478–482. [Google Scholar]
- Yin, X.-y.; Landay, M.F.; Han, W.; Levitan, E.S.; Watkins, S.C.; Levenson, R.M.; Farkas, D.L.; Prochownik, E.V. Dynamic in vivo interactions among Myc network members. Oncogene 2001, 20, 4650–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, D.L.; Ballou, B.T.; Fisher, G.W.; Fishman, D.; Garini, Y.; Niu, W.; Wachman, E.S. Microscopic and mesoscopic spectral bio-imaging. Proc. SPIE 1996, 2678, 200–209. [Google Scholar]
- Xiong, Y.; Zhao, T.; Talavera, D.; Sierra-Honigmann, R.; Farkas, D.L. A novel node-structural map for angiogenesis analysis. Medical Imaging: Physiology, Function, and Structure from Medical Images. Proc. SPIE 2005, 5747, 537–545. [Google Scholar]
- Talavera-Adame, D.; Xiong, Y.; Zhao, T.; Arias, A.E.; Sierra-Honigmann, M.R.; Farkas, D.L. Quantitative and morphometric evaluation of the angiogenic effects of leptin. J. Biomed. Opt. 2008, 13, 064017. [Google Scholar] [CrossRef] [Green Version]
- Talavera, D.; Dafoe, D.C.; Wu, G.; Klein, A.S.; Arias, A.E.; Sierra-Honigmann, R.M.; Wachsmann-Hogiu, S.; Castillo-Henkel, C.; Farkas, D.L. Enhancement of embryonic stem cell differentiation is promoted by avian chorioallantoic membranes. Tissue Eng. A 2009, 15, 3193–3200. [Google Scholar] [CrossRef]
- Krakow, D.; Robertson, S.P.; King, L.M.; Morgan, T.; Sebald, E.T.; Bertolotto, C.; Wachsmann-Hogiu, S.; Acuna, D.; Shapiro, S.S.; Takafuta, T.; et al. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation, and skeletogenesis. Nat. Genet. 2004, 36, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Wachsmann-Hogiu, S.; Krakow, D.; Sebald, E.T.; Bertolotto, C.; Acuna, D.; Farkas, D.L. Confocal and two-photon imaging in cartilage: Expression patterns of Filamin A and B. Prog. Biomed. Opt. Imaging 2004, 5, 140–145. [Google Scholar]
- Wachsmann-Hogiu, S.; Kirilova, V.; Krakow, D.; Farkas, D.L. Multiphoton, confocal and spectral microscopy for molecular imaging in cartilage. Prog. Biomed. Opt. Imaging 2005, 6, 75–81. [Google Scholar]
- Farkas, D.L.; Ballou, B.T.; Fisher, G.W.; Taylor, D.L. From in vitro to in vivo by dynamic multiwavelength imaging. Proc. SPIE 1995, 2386, 138–149. [Google Scholar]
- Ballou, B.; Fisher, G.W.; Waggoner, A.S.; Farkas, D.L.; Reiland, J.M.; Jaffe, R.; Mujumdar, R.; Mujumdar, S.; Hakala, T.R. Tumor location in vivo using cyanine fluorochrome-labeled monoclonal antibodies. Cancer Immunol. Immunother. 1995, 41, 251–263. [Google Scholar] [CrossRef]
- Ballou, B.; Fisher, G.W.; Deng, J.S.; Reiland, J.M.; Hakala, T.R.; Srivastava, M.; Farkas, D.L. Three-dimensional imaging of nucleolin trafficking in normal cells, transfectants, and heterokaryons. Proc. SPIE 1996, 2680, 124–131. [Google Scholar]
- Ballou, B.T.; Fisher, G.W.; Hakala, T.; Farkas, D.L. Tumor visualization using fluorochrome-labeled antibodies. Biotechnol. Prog. 1997, 13, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Ballou, B.T.; Fisher, G.W.; Hakala, T.R.; Farkas, D.L. Fluorochromes for tumor imaging in vivo. Cancer Detect. Prev. 1998, 22, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Farkas, D.L.; Ballou, B.; Du, C.; Fisher, G.W.; Lau, C.; Niu, W.; Wachman, E.S.; Levenson, R.M. Optical image acquisition, analysis and processing for biomedical applications. Springer Lect. Notes Comput. Sci. 1997, 1311, 663–671. [Google Scholar]
- Weissleder, R. Molecular imaging in cancer. Science 2006, 312, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Horan, P.K.; Slezak, S.E. Stable cell membrane labeling. Nature 1989, 340, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Moffatt-Blue, C.; Equils, O.; Fujita, M.; Jeong, J.; Khazenzon, N.M.; Lindsley, E.; Ljubimova, J.; Nowatzyk, A.G.; Farkas, D.L.; et al. Multimode optical imaging of small animals: Development and applications. Prog. Biomed. Opt. Imaging 2007, 8, 644105. [Google Scholar]
- Wachsmann-Hogiu, S.; Hwang, J.Y.; Lindsley, E.; Farkas, D.L. Wide-field 2-photon microscopy: Features and advantages for biomedical applications. Prog. Biomed. Opt. Imaging 2007, 8, 64411B. [Google Scholar]
- Hwang, J.Y.; Agadjanian, H.; Medina-Kauwe, L.K.; Gross, Z.; Gray, H.B.; Sorasaenee, K.; Farkas, D.L. Large field of view scanning fluorescence lifetime imaging system for multi-mode optical imaging of small animals. Prog. Biomed. Opt. Imaging 2008, 6859, 68590G. [Google Scholar]
- Hwang, J.Y. Development of a Multimode Optical Imaging System for Preclinical Applications In Vivo. Ph.D. Thesis, University of Southern California, Los Angeles, CA, USA, 2009. [Google Scholar]
- Agadjanian, H.; Jun, M.; Rentsendorj, A.; Valluripalli, V.; Hwang, J.Y.; Mahammed, A.; Marom, A.; Farkas, D.L.; Gray, H.; Gross, Z.; et al. Tumor detection and elimination by a targeted gallium corrole. Proc. Natl. Acad. Sci. USA 2009, 106, 6105–6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.Y.; Lubow, J.; Chu, D.; Gross, Z.; Gray, H.B.; Farkas, D.L.; Medina-Kauwe, L. Investigating the photosensitizer-potential of targeted gallium corrole using multimode optical imaging. Prog. Biomed. Opt. Imaging 2011, 7886, 1–6. [Google Scholar]
- Hwang, J.Y.; Gray, H.B.; Gross, Z.; Medina-Kauwe, L.; Farkas, D.L. Multimode optical imaging for translational chemotherapy: In vivo tumor detection and delineation by targeted gallium corroles. Prog. Biomed. Opt. Imaging 2011, 7902, 1–8. [Google Scholar]
- Hwang, J.Y.; Wachsmann-Hogiu, S.; Ramanujan, V.K.; Nowatzyk, A.G.; Koronyo, Y.; Medina-Kauwe, L.K.; Gross, Z.; Gray, H.B.; Farkas, D.L. Multimodal wide-field two-photon excitation imaging: Characterization of the technique for in vivo applications. Biomed. Opt. Express 2011, 2, 356–364. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Gross, Z.; Gray, H.B.; Medina-Kauwe, L.K.; Farkas, D.L. Ratiometric spectral imaging for fast tumor detection and chemotherapy monitoring in vivo. J. Biomed. Opt. 2011, 16, 066007. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Lubow, J.; Chu, D.; Agadjanian, H.; Gray, H.B.; Gross, Z.; Farkas, D.L.; Medina-Kauwe, L.K. A mechanistic study of tumor-tageted corrole toxicity. Mol. Pharm. 2011, 8, 2233–2243. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Wachsmann-Hogiu, S.; Ramanujan, V.K.; Ljubimova, J.; Gross, Z.; Gray, H.B.; Medina-Kauwe, L.K.; Farkas, D.L. A Multimode Optical Imaging System for Preclinical Applications In Vivo: Technology Development, Multi-scale Imaging and Chemotherapy Assessment. Mol. Imaging Biol. 2011, 14, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Park, J.; Kang, B.J.; Lubow, J.; Chu, D.; Farkas, D.L.; Shung, K.K.; Medina-Kauwe, L.K. Multimodality imaging in vivo for preclinical assessment of tumor-targeted doxorubicin nanoparticles. PLoS ONE 2012, 7, e34463. [Google Scholar] [CrossRef] [Green Version]
- Agadjanian, H.; Chu, D.; Hwang, J.Y.; Wachsmann-Hogiu, S.; Retsendorj, A.; Song, L.; Valluripalli, V.; Lubow, J.; Ma, J.; Sharifi, B.; et al. Chemotherapy targeting by DNA capture in viral protein particles. Nanomedicine 2012, 7, 335–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.Y.; Lubow, J.; Sims, J.; Gray, H.B.; Mahammed, A.; Gross, Z.; Medina-Kauwe, L.K.; Farkas, D.L. Investigating photoexcitation-induced mitochondrial damage by chemotherapeutic corroles using multimode optical imaging. J. Biomed. Opt. 2012, 17, 015003. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Lubow, D.J.; Ch, D.; Sims, J.; Alonso-Valenteen, F.; Gray, H.B.; Gross, Z.; Farkas, D.L.; Medina-Kauwe, L.K. Photoexcitation of tumor-targeted corroles induces singlet oxygen-mediated augmentation of cytotoxicity. J. Control. Release 2012, 163, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Farkas, D.L.; Medina-Kauwe, L.K. Analysis of targeted viral protein nanoparticles delivered to HER2+ tumors. J. Vis. Exp. 2013, 76. Available online: http://www.jove.com/video/50396?status=a52402k (accessed on 11 April 2021). [CrossRef] [PubMed]
- Ljubimova, J.Y.; Fujita, M.; Lee, B.S.; Khazenzon, N.M.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Holler, E. Nanoconjugates of poly(malic acid) with functional modules for drug delivery. NSTI Nanotech. 2006, 2, 354–357. [Google Scholar]
- Lee, B.S.; Fujita, M.; Khazenzon, N.M.; Wawrowsky, K.A.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Ljubimova, J.Y.; Holler, E. Polycefin, a new prototype of a multifunctional nanoconjugate based on poly(beta-Lmalic acid) for drug delivery. Bioconjug. Chem. 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubimova, J.Y.; Fujita, M.; Khazenzon, N.M.; Lee, B.-S.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Holler, E. Nanoconjugate based on polymalic acid for tumor targeting. Chem. Biol. Interact. 2007, 171, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.; Wachsmann, S.; Zhao, T.; Xiong, Y.; Joseph, A.; Farkas, D.L. Advanced optical imaging requiring no contrast agents—A new armamentarium for medicine and surgery. Curr. Surg. 2005, 62, 365–370. [Google Scholar] [CrossRef]
- Chung, A.; Gaon, M.; Jeong, J.; Karlan, S.; Lindsley, E.; Wachsmann-Hogiu, S.; Zhao, T.; Xiong, Y.; Farkas, D.L. Spectral imaging detects breast cancer in fresh unstained specimens. Prog. Biomed. Opt. Imaging 2006, 7, 608806. [Google Scholar]
- Chung, A.; Lindsley, E.; Wachsmann-Hogiu, S.; Farkas, D.L. In vivo cytometry—A spectrum of possibilities. Cytometry 2006, 69A, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Frykman, P.K.; Gaon, M.; Chung, A.P.; Lindsley, E.H.; Hwang, J.Y.; Farkas, D.L. Intelligent spectral signature bio-imaging in vivo for surgical applications. Progr. Biomed. Opt. Imaging 2007, 8, 64411N. [Google Scholar]
- Nyirenda, N.; Farkas, D.L.; Ramanujan, V.K. Preclinical evaluation of nuclear morphometry and tissue topology for breast carcinoma detection and margin assessment. Breast Cancer Res. Treat. 2010, 126, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanujan, V.K.; Ren, S.; Park, S.; Farkas, D.L. Non-invasive, contrast-enhanced spectral imaging of breast cancer signatures in preclinical animal models in vivo. J. Cell Sci. Ther. 2010, 1, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Swanson, E.A.; Lin, C.P.; Schuman, J.S.; Stinson, W.G.; Chang, W.; Hee, M.R.; Flotte, T.; Gregory, K.; Puliafito, C.; et al. Optical coherence tomography. Science 1991, 254, 1178–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Farkas, D.L. In vivo imaging of biological tissues using 1.3 µm optical coherence tomography. Proc. SPIE 1997, 2983, 93–100. [Google Scholar]
- Pan, Y.; Farkas, D.L. Non-invasive imaging of biological tissue with dual-wavelength OCT. Proc. SPIE 1998, 3260, 141–149. [Google Scholar]
- Pan, Y.; Farkas, D.L. Non-invasive imaging of living human skin with dual-wavelength Optical Coherence Tomography in two and three dimensions. J. Biomed. Opt. 1998, 3, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Lavelle, J.P.; Meyers, S.; Pirtskhalaishvili, G.; Bastacky, S.I.; Zeidel, M.; Farkas, D.L. High fidelity Optical Coherence Tomography of tumorigenesis in rat bladders induced by N-Methyl-N-NitrosoUrea instillation. Med Phys. 2001, 28, 2432–2440. [Google Scholar] [CrossRef]
- Frykman, P.K.; Gaon, M.; Lindsley, E.; Lechago, J.; Chung, A.P.; Xiong, Y.; Farkas, D.L. A novel, rapid, and accurate method for determining the level of aganglionosis in Hirschsprung’s Disease using spectral bioimaging. Proc. IPEG 2006, ET001, 76. [Google Scholar]
- Frykman, P.K.; Lindsley, E.H.; Gaon, M.; Farkas, D.L. Spectral imaging for precise surgical intervention in Hirschsprung’s disease. J. Biophotonics 2008, 1, 97–103. [Google Scholar] [CrossRef]
- Frykman, P.K.; Farkas, D.L. Spectral Imaging Device for Hirschsprung’s Disease. U.S. Patent Application 20100130871A1, 2 April 2008. [Google Scholar]
- Farkas, D.L.; Fisher, G.W.; Schmidt, G.; Pillai, M.; Ildstad, S. Bone marrow transplant facilitating cells-tracking and characterization by optical imaging. Tissue Eng. 1996, 69–70. [Google Scholar]
- Askenasy, N.; Farkas, D.L. Antigen barriers or available space do not restrict in situ adhesion of hematopoietic cells to bone marrow stroma. Stem Cells 2008, 20, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Askenasy, N.; Zorina, T.; Farkas, D.L.; Shalit, I. Transplanted hematopoietic cells seed in clusters in recipient bone marrow in vivo. Stem Cells 2002, 20, 301–310. [Google Scholar] [CrossRef]
- Askenasy, N.; Farkas, D.L. Optical imaging of PKH-labeled hematopoietic cells in recipient bone marrow in vivo. Stem Cells 2002, 20, 501–513. [Google Scholar] [CrossRef]
- Askenasy, N.; Shirwan, H.; Wang, Z.; Farkas, D.L. Cardiac allograft acceptance after localized bone marrow transplantation by isolated limb perfusion in non-myeloablated recipients. Stem Cells 2003, 21, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Askenasy, N.; Farkas, D.L. In vivo imaging studies of the effect of recipient conditioning, donor cell phenotype and antigen disparity on homing of haematopoietic cells to the bone marrow. Br. J. Haematol. 2003, 120, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Askenasy, N.; Stein, J.; Yaniv, I.; Farkas, D.L. The topologic and chronologic patterns of hematopoietic cell seeding in host femoral bone marrow after transplantation. Biol. Blood Marrow Transpl. 2003, 9, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Askenasy, N.; Yolcu, E.S.; Shirwan, H.; Stein, J.; Yaniv, I.; Farkas, D.L. Characterization of adhesion and viability of early seeding hematopoietic cells in the host bone marrow in vivo and in situ. Exper. Hematol. 2003, 12, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, I.; Stein, J.; Farkas, D.L.; Askenasy, N. The tale of early hematopoietic cell seeding in the bone marrow niche. Stem Cells Dev. 2006, 15, 4–16. [Google Scholar] [CrossRef]
- Askenasy, N.; Stein, J.; Farkas, D.L. Imaging approaches to hematopoietic stem and progenitor cell function and engraftment. Immunol. Investig. 2007, 36, 713–738. [Google Scholar] [CrossRef] [PubMed]
- Pearl-Yafe, M.; Stein, J.; Yolcu, E.S.; Farkas, D.L.; Shirwan, H.; Yaniv, I.; Askenasy, N. Fas transduces dual apoptotic and trophic signals in hematopoietic progenitors. Stem Cells 2007, 25, 3194–3203. [Google Scholar] [CrossRef] [PubMed]
- Kaminitz, A.; Mizrahi, K.; Yaniv, I.; Farkas, D.L.; Stein, J.; Askenasy, N. Low levels of allogeneic hematopoietic chimerism reverse autoimmune insulitis in NOD mice, while syngeneic reconstitution is ineffective. J. Autoimmun. 2009, 33, 83–91. [Google Scholar] [CrossRef]
- Yarkoni, S.; Sagiv, Y.; Kaminitz, A.; Farkas, D.L.; Askenasy, N. Targeted therapy to IL-2 receptor using diphtheria toxin and caspase-3 fusion proteins modulates regulatory T cells and ameliorates inflammatory colitis. Eur. J. Immunol. 2009, 39, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, I.; Ash, S.; Farkas, D.L.; Askenasy, N.; Stein, J. Consideration of strategies for hematopoietic cell transplantation. J. Autoimmun. 2009, 33, 255–259. [Google Scholar] [CrossRef]
- Iskovich, S.; Goldenberg-Cohen, N.; Stein, J.; Yaniv, I.; Farkas, D.L.; Askenasy, N. β-Cell Neogenesis: Experimental Considerations in Adult Stem Cell Differentiation. Stem Cells Dev. 2010, 20, 569–582. [Google Scholar] [CrossRef]
- Lo Celso, C.; Wu, J.W.; Lin, C.P. In vivo imaging of hematopoietic stem cells and their microenvironment. J. Biophoton. 2009, 2, 619–631. [Google Scholar] [CrossRef] [PubMed]
- MacGowan, G.A.; Du, C.; Farkas, D.L.; Koretsky, A.P. Measurement of intracellular calcium during changes in inotropy in the isolated perfused mouse heart with rhod-2. J. Card. Fail. 1998, 4, 8–9. [Google Scholar] [CrossRef]
- Du, C.; MacGowan, G.; Farkas, D.L.; Koretsky, A.S. Calcium measurements in perfused mouse heart: Quantitating fluorescence and absorbance of Rhod-2 by application of photon migration theory. Biophys. J. 2001, 80, 549–561. [Google Scholar] [CrossRef] [Green Version]
- MacGowan, G.A.; Du, C.; Suhan, J.P.; Koretsky, A.P.; Farkas, D.L. Rhod-2 based measurements of intracellular calcium in the perfused mouse heart: Cellular and subcellular localization and response to positive inotropy. J. Biomed. Opt. 2001, 6, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; MacGowan, G.A.; Farkas, D.L.; Koretsky, A.P. Calibration of calcium dissociation constant of Rhod2 in the perfused mouse heart using manganese quenching. Cell Calcium 2001, 29, 217–227. [Google Scholar] [CrossRef]
- Wachman, E.S.; Poage, R.E.; Farkas, D.L.; Meriney, S.D. Variability in the spatial distribution of presynaptic calcium entry during single action potentials. J. Neurosci. 2004, 24, 2877–2885. [Google Scholar] [CrossRef] [Green Version]
- Shonat, R.D.; Wachman, E.S.; Niu, W.; Koretsky, A.P.; Farkas, D.L. Near-simultaneous hemoglobin saturation and oxygen tension maps in mouse brain using an AOTF microscope. Biophys. J. 1997, 73, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Shonat, R.D.; Wachman, E.S.; Niu, W.; Koretsky, A.P.; Farkas, D.L. Near-simultaneous hemoglobin saturation and oxygen tension maps in mouse cortex during amphetamine stimulation. Oxygen Transport to Tissue XX. Adv. Exp. Med. Biol. 1998, 454, 149–158. [Google Scholar] [PubMed]
- Shonat, R.D.; Kight, A.C. Oxygen tension imaging in the mouse retina. Ann. Biomed. Eng. 2003, 31, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Farkas, D.L. Molecular biology in surgical oncology: The role of molecular imaging. In Principles and Practice of Surgical Oncology —Multidisciplinary Approach to Difficult Problems; Silberman, H., Silberman, A.W., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2010; pp. 18–28. [Google Scholar]
- Jack, M.D., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Update on hypothetical model of Alzheimer’s disease biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Koronyo-Hamaoui, M.; Ko, M.H.; Azoulay, D.; Seksenyan, A.; Koronyo, Y.; Kunis, G.; Rogeri, P.; Bakhsheshian, J.; Black, K.; Farkas, D.L.; et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: Local elevation of IL-10 and MMP-9. J. Neurochem. 2009, 11, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. NeuroImage 2011, 54 (Suppl. S1), S204–S217. [Google Scholar] [CrossRef] [Green Version]
- Koronyo, Y.; Koronyo, H.M.; Black, K.; Schwartz, M.; Farkas, D.L. Optical Method for Detecting Alzheimer’s Disease by Systemic Administration of Curcumin. U.S. Patent 9,839,699, 12 December 2017. [Google Scholar]
- Koronyo, Y.; Koronyo, H.M.; Black, K.; Schwartz, M.; Farkas, D.L. Optical Method for Detecting Alzheimer’s Disease Using Curcumin. U.S. Patent 10,512,699, 24 December 2019. [Google Scholar]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. ICI Insight 2017, 2, e93621. [Google Scholar] [CrossRef] [PubMed]
- Nowatzyk, A.G. Optical Beam Scanning System Having a Synthetic Center of Beam Rotation. U.S. Patent 10,520,721, 31 December 2019. [Google Scholar]
- Nowatzyk, A.G. Low Noise Photo-Parametric Solid-State Amplifier. U.S. Patent 8,901,997, 2 December 2014. [Google Scholar]
- Kirkwood, J.M.; Farkas, D.L.; Chakraborty, A.; Dyer, K.F.; Tweardy, D.J.; Abernethy, J.L.; Edington, H.D.; Donnelly, S.S.; Becker, D. Systemic interferon-treatment leads to Stat3 inactivation in melanoma precursor lesions. Mol. Med. 1999, 5, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Farkas, D.L.; Kirkwood, J.M.; Abernethy, J.L.; Edington, H.D.; Becker, D. Macroscopic spectral imaging and gene expression analysis of the early stages of melanoma. Mol. Med. 1999, 5, 785–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valesky, M.; Spang, A.J.; Fisher, G.W.; Farkas, D.L.; Becker, D. Non-invasive, dynamic fluorescence imaging of human melanomas reveals that targeted inhibition of bFGF and FGFR-1 blocks tumor growth by inducing melanoma cell apoptosis. Mol. Med. 2002, 8, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaff-Smith, A.; Kirkwood, J.M.; Edington, H.D.; Jukic, D.M.; Farkas, D.L.; Becker, D. Fluorescence imaging analysis of upstream regulators and downstream targets of STAT3 in melanoma precursor lesions obtained from patients before and after systemic low-dose interferon-α treatment. Mol. Imaging 2003, 2, 65–73. [Google Scholar] [CrossRef]
- Farkas, D.L.; Becker, D. Applications of spectral imaging: Detection and analysis of human melanoma and its precursors. Pigment Cell Res. 2001, 14, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Joseph, A.O.N.R.; Lindsley, E.H.; Farkas, D.L. Polarization-sensitive digital dermoscopy for image processing-assisted evaluation of atypical nevi: Towards step-wise detection of melanoma. Prog. Biomed. Opt. Imaging 2011, 79020K, 1–6. [Google Scholar]
- MacKinnon, N.B.; Vasefi, F.; Gussakovsky, E.; Bearman, G.H.; Chave, R.; Farkas, D.L. In vivo skin chromophore mapping using a multimode imaging dermoscope (SkinSpect). Proc. SPIE 2013, 8587, 85870U. [Google Scholar]
- Vasefi, F.; Saager, R.B.; Durkin, A.J.; MacKinnon, N.B.; Gussakovsky, E.; Chave, R.; Farkas, D.L. Quantifying the optical properties and chromophore concentrations of turbid media using polarization sensitive hyperspectral imaging: Optical phantom studies. Proc. SPIE 2013, 8587, 1–9. [Google Scholar]
- MacKinnon, N.B.; Vasefi, F.; Farkas, D.L. Toward in-vivo diagnosis of skin cancer using multimode imaging dermoscopy: (I) Clinical system development and validation. Proc. SPIE 2014, 8947, 89470I. [Google Scholar]
- Vasefi, F.; MacKinnon, N.B.; Farkas, D.L. Toward in-vivo diagnosis of skin cancer using multimode imaging dermoscopy: (II) Molecular mapping of highly pigmented lesions. Proc. SPIE 2014, 8947, 89470J. [Google Scholar]
- Vasefi, F.; MacKinnon, N.B.; Farkas, D.L. Multimode imaging applied towards diagnosis of skin cancer (SkinSpect). Proc. Can. Med Biol. Eng. Soc. 2014, 33–40. [Google Scholar]
- Vasefi, F.; MacKinnon, N.; Farkas, D.L. Polarization-sensitive hyperspectral imaging in vivo: A multimode dermoscope for skin analysis. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasefi, F.; MacKinnon, N.B.; Saager, R.; Durkin, A.J.; Chave, R.; Farkas, D.L. Quantifying the optical properties of turbid media using polarization sensitive hyperspectral imaging (SkinSpect): Two-layer optical phantom studies. Proc. SPIE 2015, 9328, 93280A. [Google Scholar]
- Vasefi, F.; MacKinnon, N.B.; Saager, R.; Durkin, A.J.; Kelly, K.; Farkas, D.L. Quantifying the optical properties of turbid media using polarization sensitive hyperspectral imaging (SkinSpect): Two-layer optical phantom studies II. Proc. SPIE 2016, 9711, 971110. [Google Scholar]
- Vasefi, F.; MacKinnon, N.; Saager, R.; Kelly, K.M.; Maly, T.; Booth, N.; Durkin, A.J.; Farkas, D.L. Separating melanin from hemodynamics in nevi using multimode hyperspectral dermoscopy and spatial frequency domain spectroscopy. J. Biomed. Opt. 2016, 21, 114001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasefi, F.; MacKinnon, N.; Saager, R.; Kelly, K.M.; Maly, T.; Chave, R.; Booth, N.; Durkin, A.J.; Farkas, D.L. Multimode optical dermoscopy (SkinSpect) analysis for skin with melanocytic nevus. Proc. SPIE 2016, 9711, 971110. [Google Scholar]
- Burton, K.; Zelikowsky, R.; Shandling, D.; Lindsley, E.; Farkas, D.L. Contrast enhancement in biomedical optical imaging using ultrabright color LEDs. Prog. Biomed. Opt. Imaging 2007, 8, 64411I. [Google Scholar]
- Farkas, D.L.; Vasefi, F.; MacKinnon, N.B.; Durkin, A.J.; Kelly, K.M. Future of care for patients at high risk for melanoma: From multimode, hyperspectral dermoscopy to self-imaging with smartphone. Melanoma Res. 2016, 26, E31. [Google Scholar]
- MacKinnon, N.B.; Vasefi, F.; Farkas, D.L. Melanoma detection using smartphone and multimode hyperspectral imaging. Proc. SPIE 2016, 9711, 971117. [Google Scholar]
- Kim, S.; Cho, D.; Kim, J.; Kim, M.; Youn, S.; Jang, J.E.; Je, M.; Lee, D.H.; Lee, B.; Farkas, D.L.; et al. Smartphone-based multispectral imaging: System development and potential for mobile skin diagnosis. Biomed. Opt. Express 2016, 7, 5294–5307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasefi, F.; Mackinnon, N.; Booth, N.; Durkin, A.J.; Kelly, K.; Farkas, D.L. Melanoma surveillance by multimode, hyperspectral dermoscopy and self-imaging using smartphone in high-risk patients. J. Am. Acad. Dermatol. 2017, 76, AB168. [Google Scholar]
- Vasefi, F.; Mackinnon, N.; Horita, T.; Shi, K.; Munia, T.T.; Tavakolian, K.; Farkas, D.L.; Rezai, R.F. A smartphone application for psoriasis management. J. Am. Acad. Dermatol. 2017, 76, AB27. [Google Scholar]
- Farkas, D.L.; MacKinnon, N.; Vasefi, F. Method and System for Characterizing Tissue in Three Dimensions using Multimode Optical Measurements. U.S. Patent Application No. US20150374309A1, 31 July 2015. [Google Scholar]
- Farkas, D.L.; MacKinnon, N.; Vasefi, F. Disposable Calibration End-Cap for Use in a Dermoscope and Other Optical Instruments. U.S. Patent Application No. US20150018645A1, 15 July 2014. [Google Scholar]
- Vasefi, F.; Mackinnon, N.; Farkas, D.L. Hyperspectral and Multispectral Imaging in Dermatology. In Imaging in Dermatology; Hamblin, M.R., Avci, P., Gupta, G.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 187–220. [Google Scholar]
- Vasefi, F.; Booth, N.; Hafizi, H.; Farkas, D.L. Multimode Hyperspectral Imaging for Food Quality and Safety. In Hyperspectral Imaging in Agriculture, Food and Environment; InTech Open: London, UK, 2018; pp. 11–26. [Google Scholar]
- Gerstner, A.O.H.; Laffers, W.; Farkas, D.L.; Martin, R.; Bendix, J.; Thies, B. Hyperspectral imaging of mucosal surfaces in patients. J. Biophotonics 2012, 5, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Regeling, B.; Laffers, W.; Gerstner, A.O.; Westermann, S.; Muller, N.A.; Schmidt, K.; Bendix, J.; Thies, B. Development of an image pre-processor for operational hyperspectral laryngeal cancer detection. J. Biophotonics 2016, 9, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Eggert, D.; Bengs, M.; Westermann, S.; Gessert, N.; Gerstner, A.O.; Müller, N.A.; Schlaefer, A.; Betz, C.; Laffers, W. In vivo detection of laryngeal cancer by hyperspectral imaging combined with deep learning methods (conference presentation). In Imaging, Therapeutics, and Advanced Technology in Head and Neck Surgery and Otolaryngology; International Society for Optics and Photonics: Bellingham, WA, USA, 2020; Volume 11213, p. 112130L. [Google Scholar]
- Yoon, J.; Joseph, J.; Waterhouse, D.J.; Luthman, A.S.; Gordon, G.S.D.; di Pietro, M.; Januszewicz, W.; Fitzgerald, R.C.; Bohndiek, S.E. A clinically translatable hyperspectral endoscopy system for imaging the gastrointestinal tract. Nat. Commun. 2019, 10, 1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halicek, M.; Lu, G.; Little, J.V.; Wang, X.; Patel, M.; Griffith, C.C.; El-Deiry, M.W.; Chen, A.Y.; Fei, B. Deep convolutional neural networks for classifying head and neck cancer using hyperspectral imaging. J. Biomed. Opt. 2017, 22, 060503. [Google Scholar] [CrossRef] [PubMed]
- Grigoroiu, A.; Yoon, J.; Bohndiek, S.E. Deep learning applied to hyperspectral endoscopy for online spectral classification. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Boeriu, A.; Boeriu, C.; Drasovean, S.; Pascarenco, O.; Mocan, S.; Stoian, M.; Dobru, D. Narrow-band imaging with magnifying endoscopy for the evaluation of gastrointestinal lesions. World J. Gastrointest. Endosc. 2015, 7, 110–120. [Google Scholar] [CrossRef]
- He, Z.; Wang, P.; Ye, X. Novel endoscopic technologies in medical trial research: Recent advancements and future prospects. BioMed Eng. 2021, 20, 5. [Google Scholar]
- Mie, G. Beitrage zur Optik truber Meiden speziell kolloidaler Metallosungen. Ann. Phys. 1908, 25, 377–445. [Google Scholar] [CrossRef]
- Backman, V.; Wallace, M.B.; Perelman, L.T.; Arendt, J.T.; Gurjar, R.; Müller, M.G.; Zhang, Q.; Zonios, G.; Kline, E.; McGilligan, J.A.; et al. Detection of preinvasive cancer cells. Nature 2000, 406, 35–36. [Google Scholar] [CrossRef]
- Lindsley, E.H. Endoscopic Imaging Elastic Scattering Spectroscopy for In Vivo Detection of Lung Cancer. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2005. [Google Scholar]
- Lindsley, E.; Wachman, E.S.; Farkas, D.L. The Hyperspectral Imaging Endoscope: A new tool for in vivo cancer detection. Prog. Biomed. Opt. Imaging 2004, 5, 75–82. [Google Scholar]
- Lindsley, E.H.; Farkas, D.L. Quantitative Endoscopic Imaging Elastic Scattering Spectroscopy: 1. Model System/Tissue Phantom Validation. Prog. Biomed. Opt. Imaging 2008, 6870, 68700I. [Google Scholar]
- Farkas, D.L.; Wachman, E.S.; Wachman, J.; Lindsley, E.; Farkas, M. Imaging Elastic Scattering Spectral Endoscopy. U.S. Patent 7,428,048, 23 September 2008. [Google Scholar]
- Stolzfus, C.; Barbour, R.; Atherton, D.; Barber, Z. Micro-sized tunable liquid crystal optical filters. Opt. Lett. 2017, 42, 2090–2093. [Google Scholar] [CrossRef]
- Vasefi, F.; Barbour, R.; Stolzfus, C.; Barber, Z.; Farkas, D.L. Mie scattering characterization by ultra-spectral illumination using micro-sized tunable liquid crystal optical filters. Proc. SPIE 2018, 10497, 104971D. [Google Scholar]
- Vasefi, F.; MacKinnon, N.; Farkas, D.L.; Kateb, B. Review of the potential of optical technologies for cancer diagnosis in neurosurgery: A step toward intraoperative neurophotonics. Neurophotonics 2017, 4, 011010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zellweger, M.; Goujon, D.; Conde, R.; Forrer, M.; van den Bergh, H.; Wagnieres, G. Absolute autofluorescence spectra of human healthy, metaplastic, and early cancerous bronchial tissue in vivo. Appl. Opt. 2001, 40, 3784–3791. [Google Scholar] [CrossRef]
- Joseph, A.O.N. Hyperspectral Imaging for Detection, Diagnosis and Staging of Cancer. Ph.D. Thesis, University of Southern California, Los Angeles, CA, USA, 2012. [Google Scholar]
- Carver, G.E.; Farkas, D.L.; Porque, J.; Feder, K.S.; Westbrook, P.S. Visible Wavelength Fiber Bragg Grating Arrays for High-Speed Biomedical Spectral Sensing In Bragg Gratings, Photosensitivity, and Poling in Glass Waveguides; OSA Technical Digest; Optical Society of America: Washington, DC, USA, 2010. [Google Scholar]
- Carver, G.E.; Locknar, S.A.; Morrison, W.A.; Farkas, D.L. High-speed multispectral confocal imaging. Proc. SPIE 2013, 8587, 1–10. [Google Scholar]
- Carver, G.E.; Locknar, S.A.; Morrison, W.A.; Farkas, D.L. Multispectral imaging for diagnosis and treatment. Proc. SPIE 2014, 8947, 89470L. [Google Scholar]
- Carver, G.E.; Locknar, S.A.; Morrison, W.A.; Ramanujan, V.K.; Farkas, D.L. High-speed multispectral confocal imaging. J. Biomed. Opt. 2014, 19, 36016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, G.E.; Locknar, S.A.; Weaver, D.L.; Stein, J.L.; Stein, G.S. Real-time detection of breast cancer at the cellular level. J. Cell Physiol. 2019, 234, 5413–5419. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Zhao, M.; Nowatzyk, A.; Farkas, D.L. Intelligent non-contact surgeon-computer interface using hand gesture recognition. In Three-Dimensional Image Capture and Applications; Corner, B.D., Mochimaru, M., Sitnik, R., Eds.; International Society for Optics and Photonics: Bellingham, WA, USA, 2008; Volume 6805. [Google Scholar]
- Gawande, A. The Checklist Manifesto: How to Get Things Right; Metropolitan Books: New York, NY, USA, 2009; 240p. [Google Scholar]
- Thomas, L. Recent Advances in Robotic Surgery. Available online: www.news-medical.net/health/Recent-Advances-in-Robotic-Surgery.aspx (accessed on 11 April 2021).
- Farkas, D.L.; Leif, R.; Nicolau, D. Believing in Seeing, vingt ans après. Prog. Biomed. Opt. Imaging 2011, 7902, xiii–xiv. [Google Scholar]
- Adler, S.N. The history of time for capsule endoscopy. Ann. Transl. Med. 2017, 5, 194. [Google Scholar] [CrossRef] [Green Version]
- Farkas, D.L. (Ed.) Spectral Imaging in Biology, Medicine and Surgery; Springer: Berlin/Heidelberg, Germany, 2022; in preparation. [Google Scholar]
- Reason, J. Human error: Models and management. Br. Med. J. 2000, 320, 768–770. [Google Scholar] [CrossRef] [Green Version]
- Mardian, Y.; Kosasih, H.; Karyana, M.; Neal, A.; Lau, C.-Y. Review of current COVID-19 diagnostics and opportunities for further development. Front. Med. 2021, 8, 615099. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Sang, Z.; Kim, Y.J.; Xiang, Y.; Cohen, T.; Belford, A.K.; Huet, A.; Conway, J.F.; Sun, J.; Taylor, D.J.; et al. Potent neutralizing nanobodies resist convergent circulating variants of SARS-CoV-2 by targeting diverse and conserved epitopes. Nat. Commun. 2021, 12, 4676. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Baker, M.A.B.; Hong, M.; Jin, D. The Quest for Optical Multiplexing in Bio-discoveries. Chem 2018, 4, 997–1021. [Google Scholar] [CrossRef] [Green Version]
- De Puig, H.; Lee, R.A.; Najjar, D.; Tan, X.; Soekensen, L.R.; Angenent-Mari, N.M.; Donghia, N.M.; Weckman, N.E.; Ory, A.; Ng, C.F.; et al. Minimally instrumented SHERLOCK (miSHERLOCK) for CRISPR-based point-of-care diagnosis of SARS-CoV-2 and emerging variants. Sci. Adv. 2021, 7, eabh2944. [Google Scholar] [CrossRef] [PubMed]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Combined Imaging Modes | Information Obtained |
|---|---|---|
| •Nanoconstruct therapy | Fluorescence intensity Spectral imaging | 1. Dynamic monitoring of nanoconstruct distribution and examination of tumor targeting capability 2. Examination of nanoconstruct clearance 3. Nanoconstruct effects on specific organs |
| •HerGa chemotherapy | Fluorescence intensity Spectral and lifetime imaging Scanning/full field two photon- excited fluorescence imaging | 1. Dynamic monitoring of HerGa and S2Ga distributions and tumor targeting capability assessment 2. Examination of HerGa accumulation kinetics 3. Mechanism of HerGa action and effects on organs 4. Tumor environment information 5. Usefulness of HerGa in surgical tumor detection |
| Imaging Mode | Biomedical Application | Capability Derived |
| •Fluorescence intensity | Measurement of relative accumulated concentration and discrimination of 2 fluorophores (AF680 and Rh123) conjugated to drug molecules in nude mice | Kinetics/Dynamics |
| •Spectral imaging | Quantitative discrimination of fluorophores (fluorescein and corroles) from autofluorescence in nude mice | Quantification |
| •Fluorescence lifetime imaging | Monitoring functional status in vicinity of fluorophores and quantitative discrimination between them | Environment assessment Quantification |
| •Intravital confocal imaging | Observation of microstructures such as muscle fibers and blood vessels without biopsy or staining | High resolution/ magnification |
| •Scanning/widefield 2-photon excited fluorescence imaging | High magnification/resolution observation of intact tissues (tumor regions) inside small animals in vivo and ex vivo (tumors, livers, eyeballs of AD mice) | High resolution/ magnification |
| •Bioluminescence imaging | Detection of ATP and enzymatic activity in engineered nude mice | High sensitivity |
| Sensitivity (%) | Specificity (%) | Mode | References | |
|---|---|---|---|---|
| 1. Cytopathology ** (Papanicolau test) | 97 | 99 | S | [65,66,67,68] |
| 2. Histopathology ** (H&E, immunohistochem.) | 98 | 96 | S | [59,60,61,62,63,64,165,166,167] |
| 3. Tissue oxygenation mapping * | 100 | 100 | M2 | [73,150,151] |
| 4. Breast cancer * (no contrast agent) Note: Second set with more advanced analysis. Note: With multimode acquisition/analysis | 91 96 96 | 86 92 97 | S S M7 | [117] [117] [116,118] |
| 5. Lymph nodes in vivo * (BC metastasis assess.) | 100 | 100 | M4 | [120,121] |
| 6. Hirschsprung’s Disease in vivo * Note: Second set with more advanced analysis | 97 98 | 94 98 | S S | [127,128,129] [128] |
| 7. Alzheimer’s Disease *,** | 100 | 100 | M3 | [156,157,158,159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farkas, D.L. Biomedical Applications of Translational Optical Imaging: From Molecules to Humans. Molecules 2021, 26, 6651. https://doi.org/10.3390/molecules26216651
Farkas DL. Biomedical Applications of Translational Optical Imaging: From Molecules to Humans. Molecules. 2021; 26(21):6651. https://doi.org/10.3390/molecules26216651
Chicago/Turabian StyleFarkas, Daniel L. 2021. "Biomedical Applications of Translational Optical Imaging: From Molecules to Humans" Molecules 26, no. 21: 6651. https://doi.org/10.3390/molecules26216651
APA StyleFarkas, D. L. (2021). Biomedical Applications of Translational Optical Imaging: From Molecules to Humans. Molecules, 26(21), 6651. https://doi.org/10.3390/molecules26216651