
The Antileukemic Effect of Xestoquinone, A Marine-Derived Polycyclic Quinone-Type Metabolite, Is Mediated through ROS-Induced Inhibition of HSP-90

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
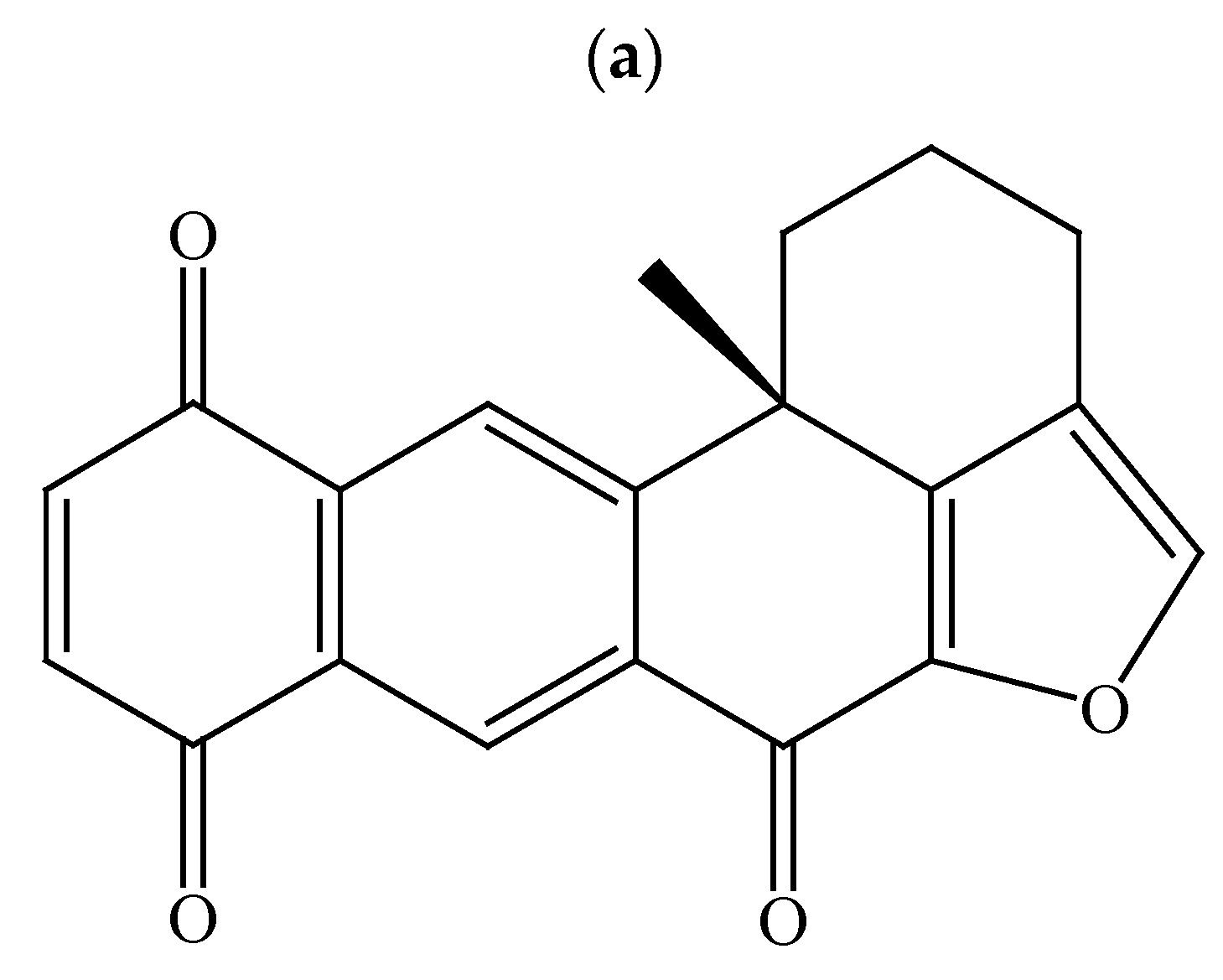
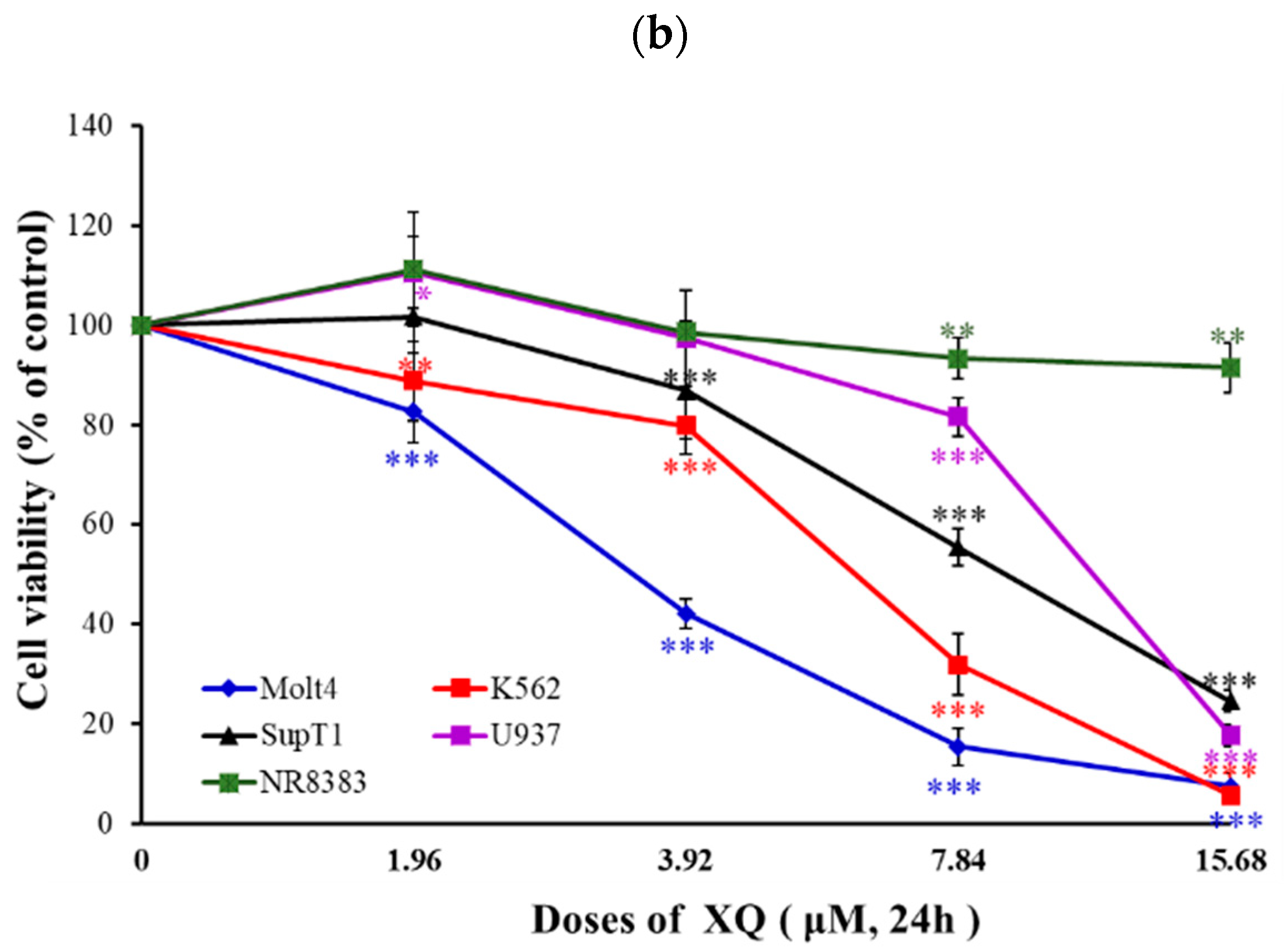
2.1. Effect of Xestoquinone on Cellular Growth of Hematologic Cancer Cells
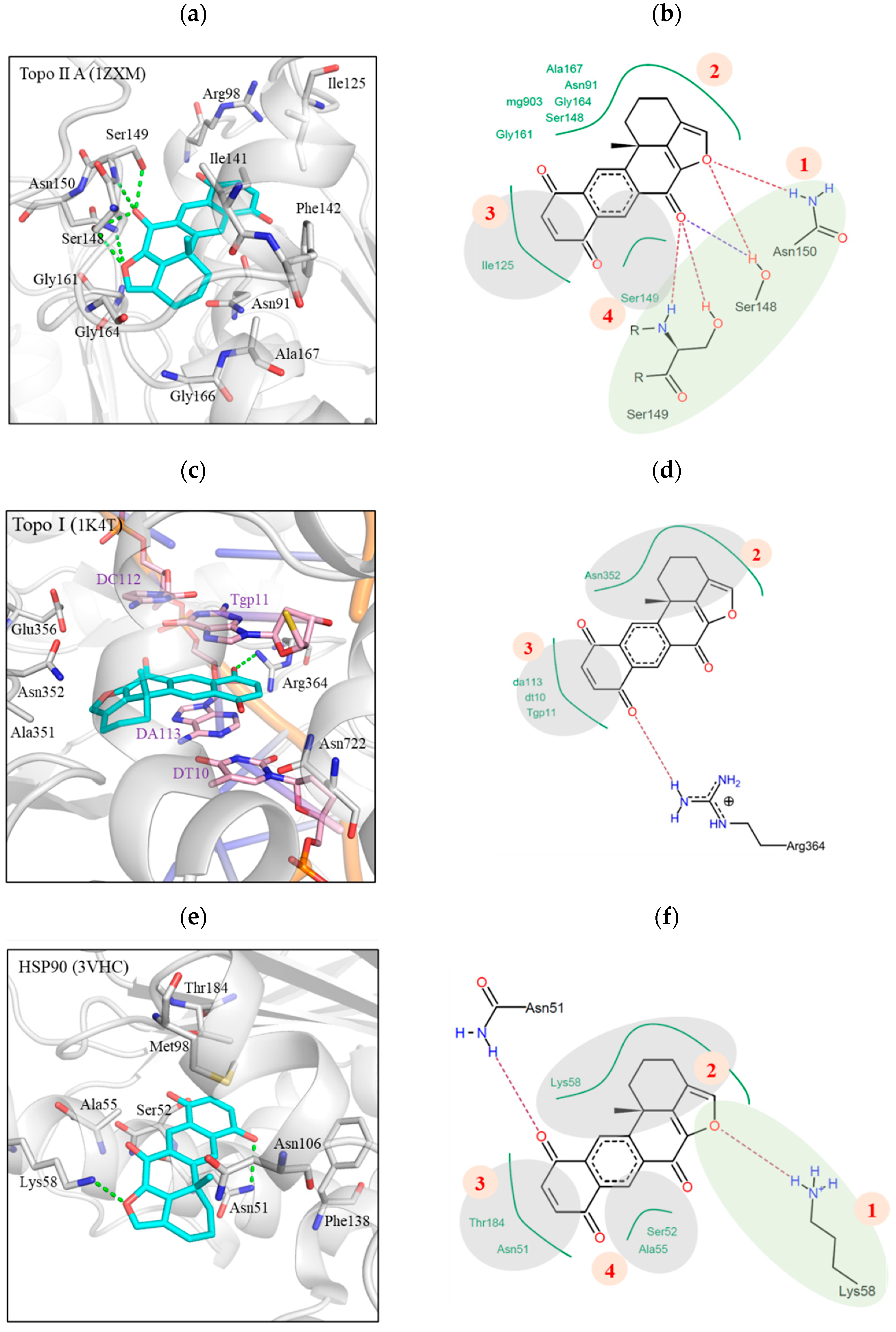
2.2. Xestoquinone Interacted with Multiple Targets including Topo I, Topo II, and HSP-90, as Demonstrated by Molecular Docking Analysis
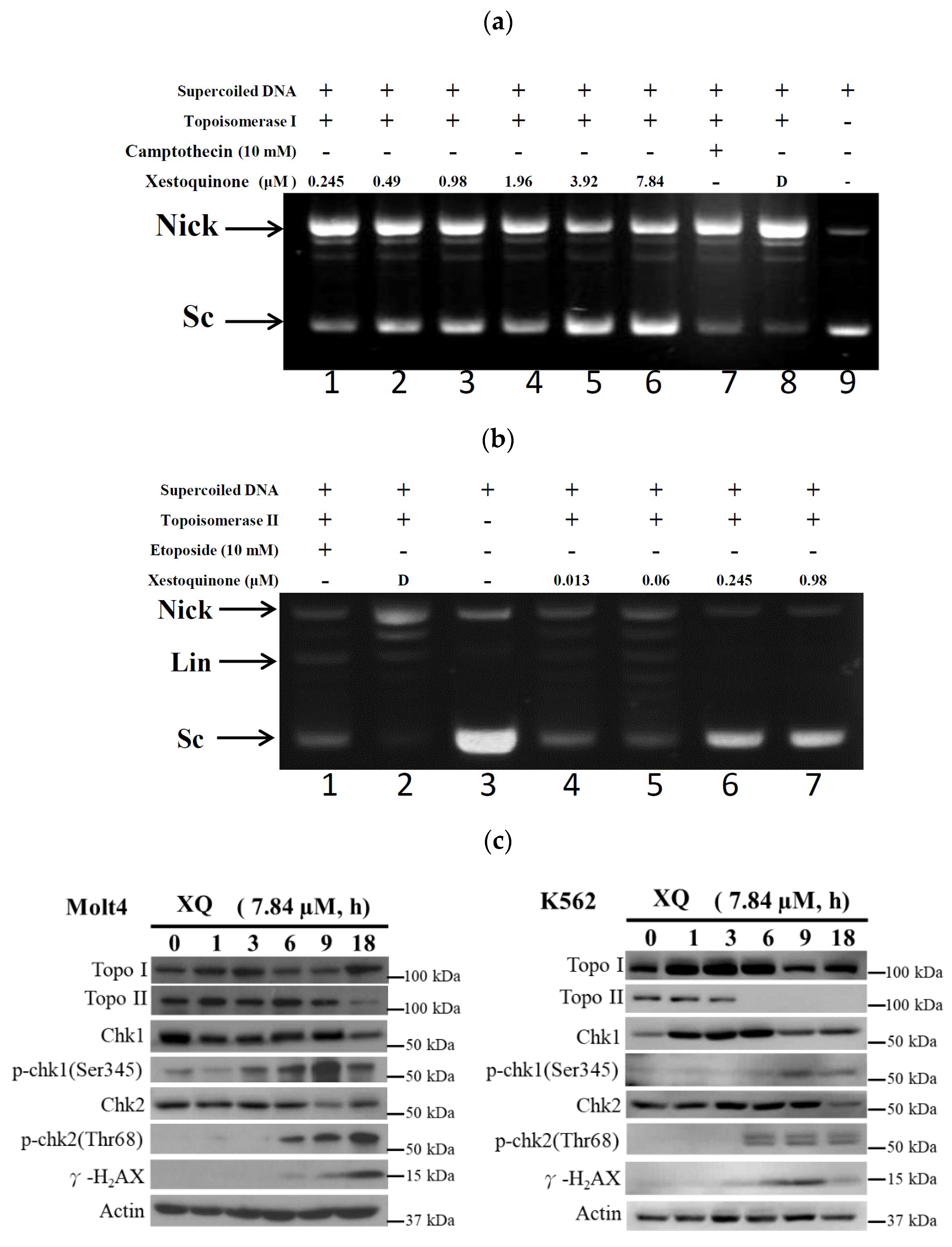
2.3. Xestoquinone Inhibited Topoisomerase I and II Activities
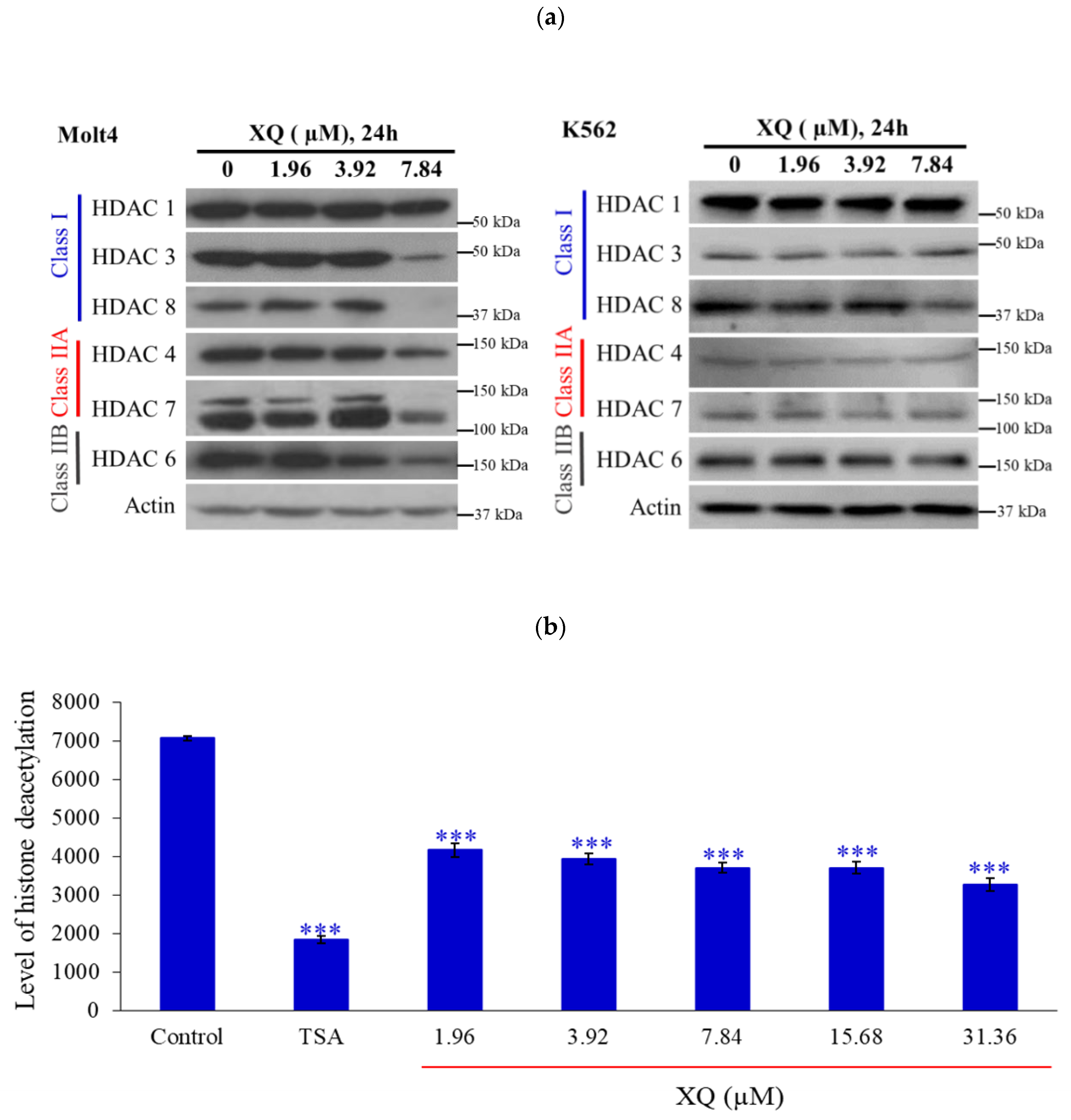
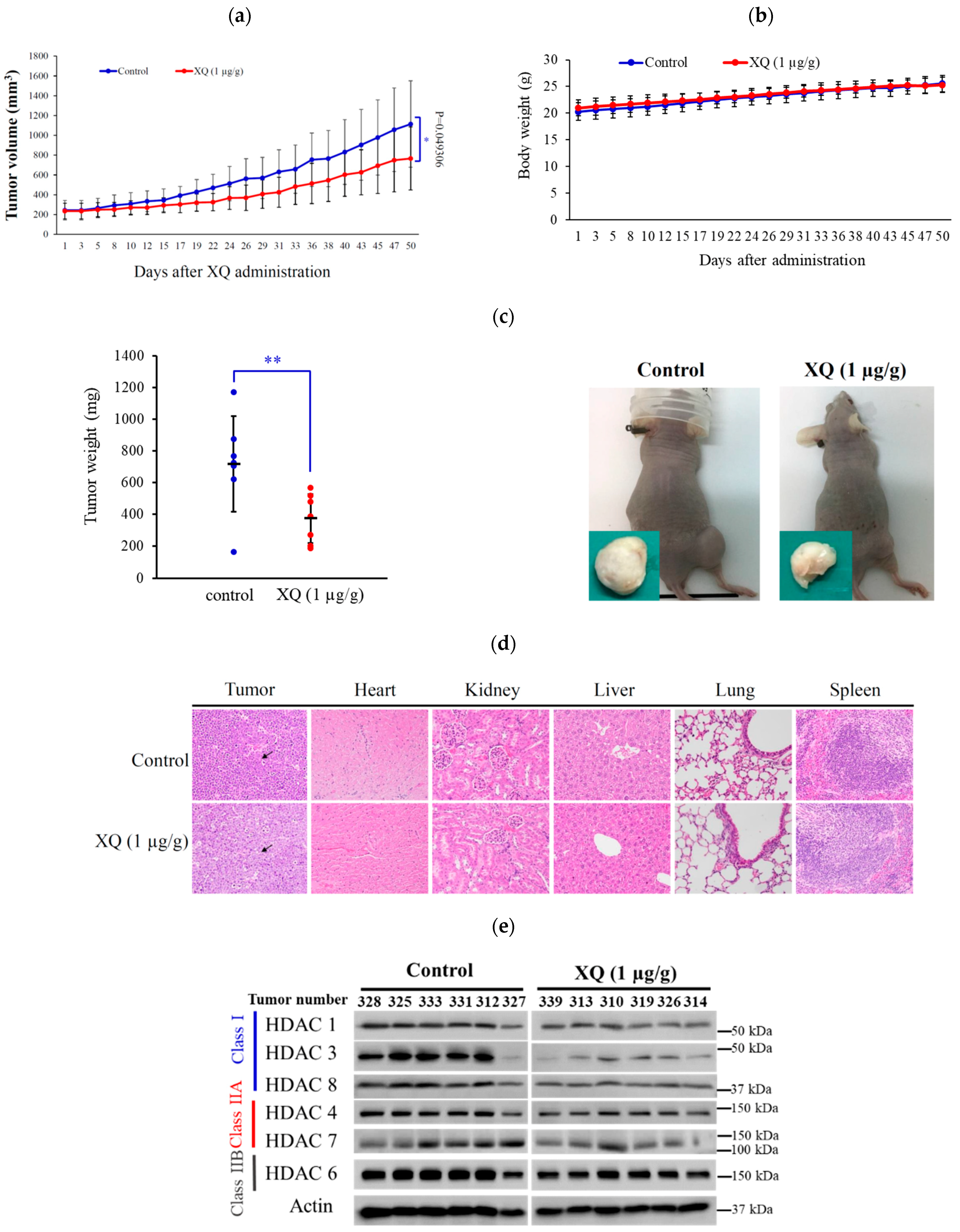
2.4. Xestoquinone Inhibited Histone Deacetylases (HDACs) in Human Leukemia Molt-4 Cells
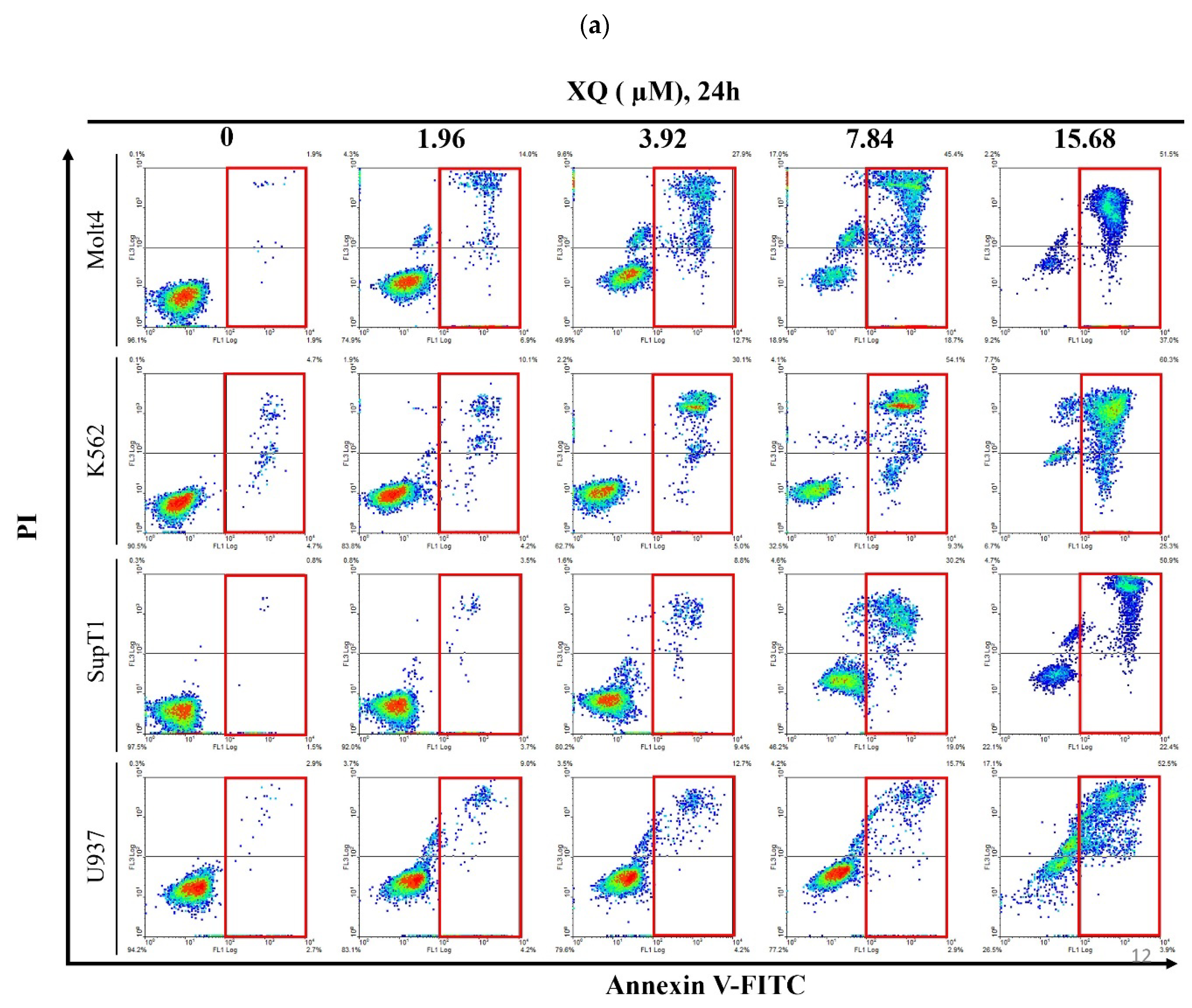
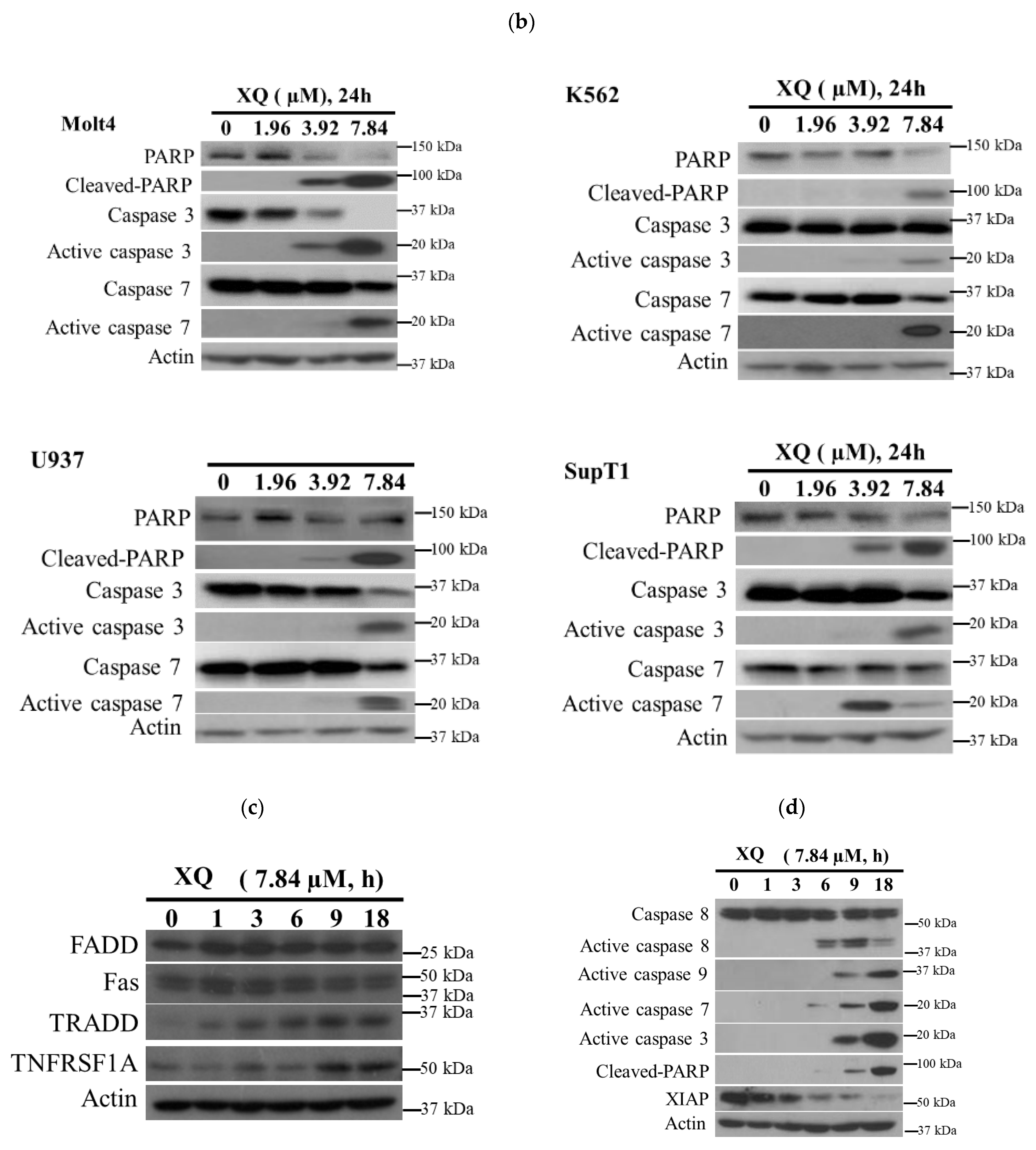
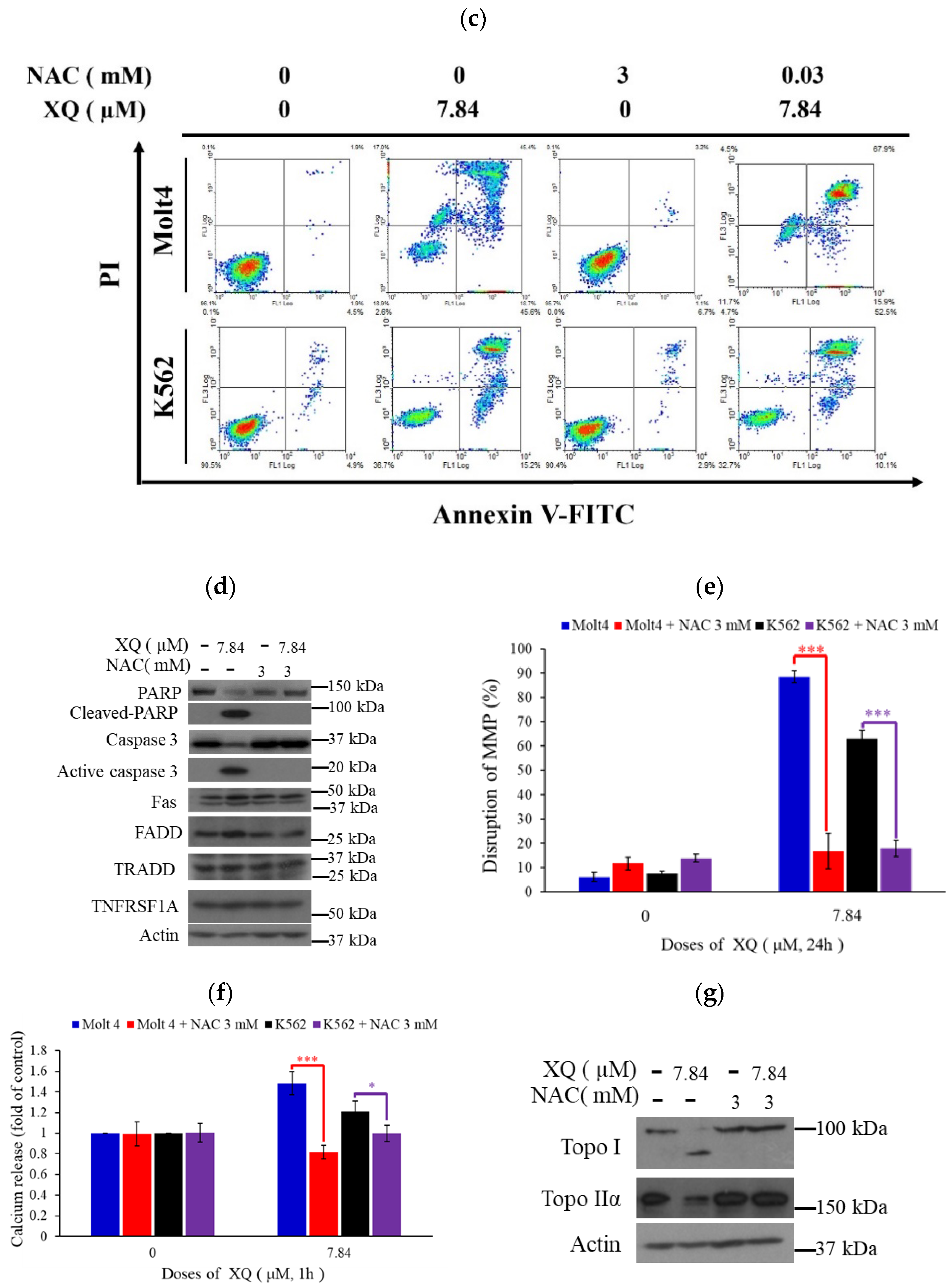
2.5. Xestoquinone Induced Cell Death in Human Leukemia Cells via Death Receptor Pathway and Apoptosis
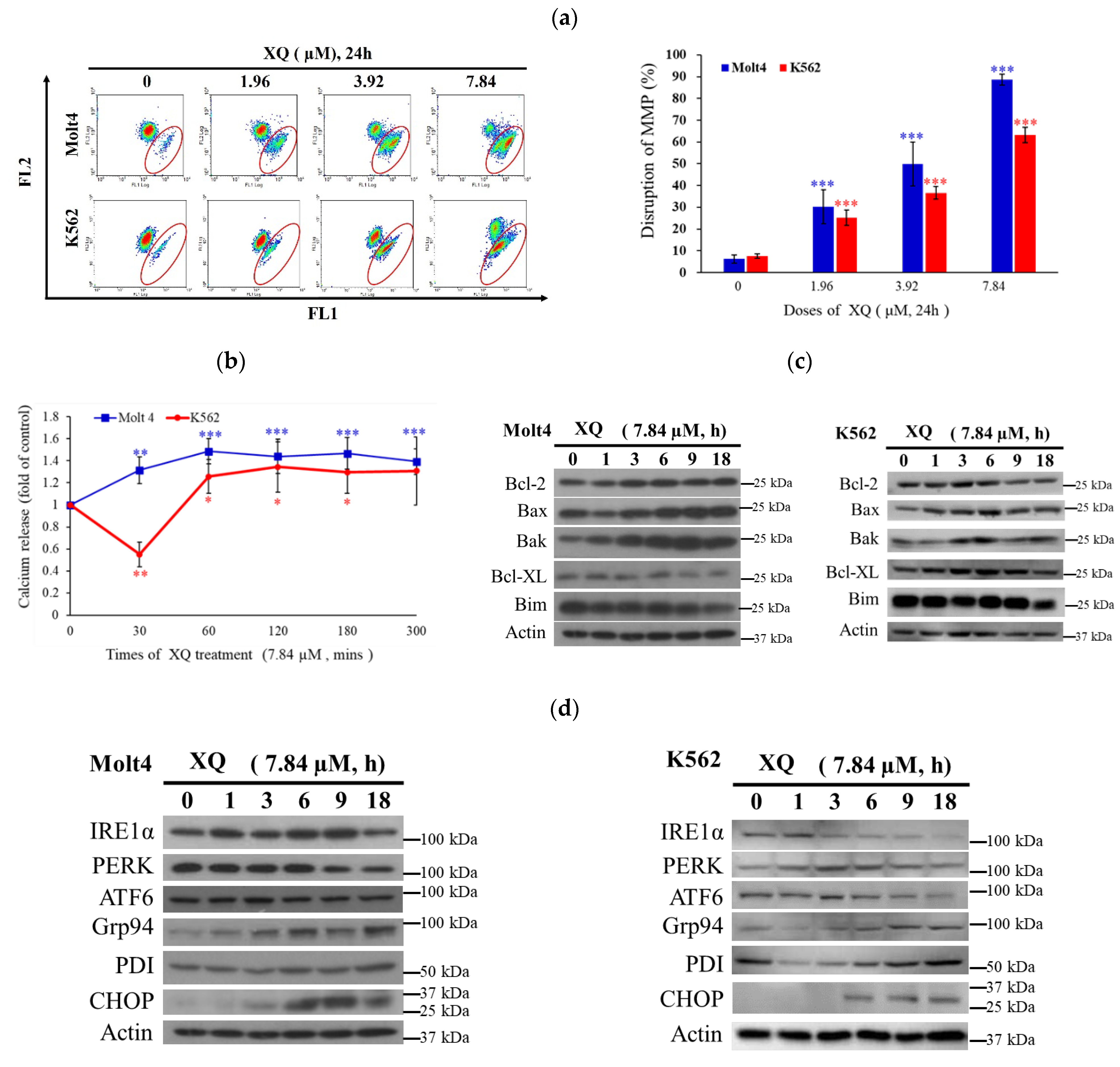
2.6. Xestoquinone Induced Mitochondrial Dysfunction, ER Stress, and Calcium Release in Human Leukemia Cells
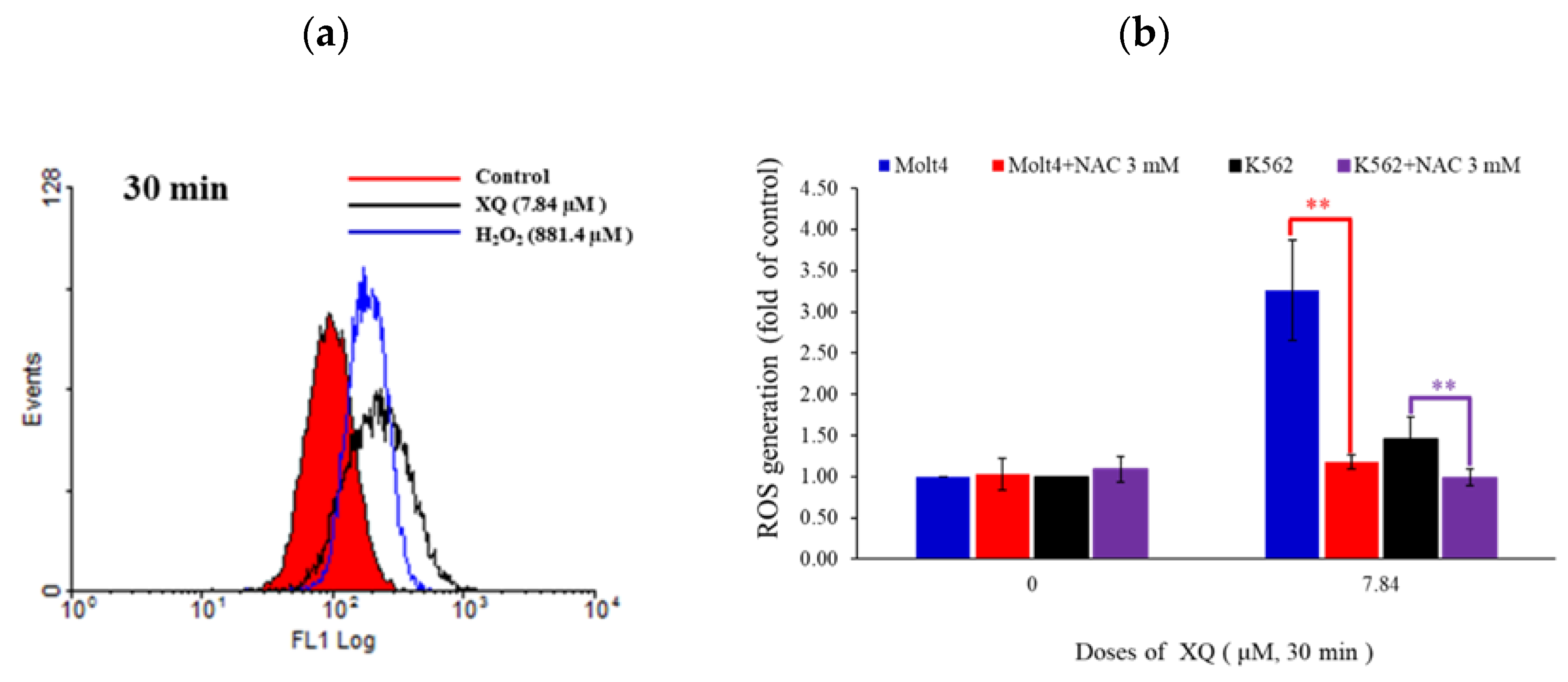
2.7. Xestoquinone Induced Topoisomerase Cleavage, Resulting in Cell Apoptosis via ROS Generation
2.8. Effect of Xestoquinone on Tumor Growth in Xenograft Animal Model
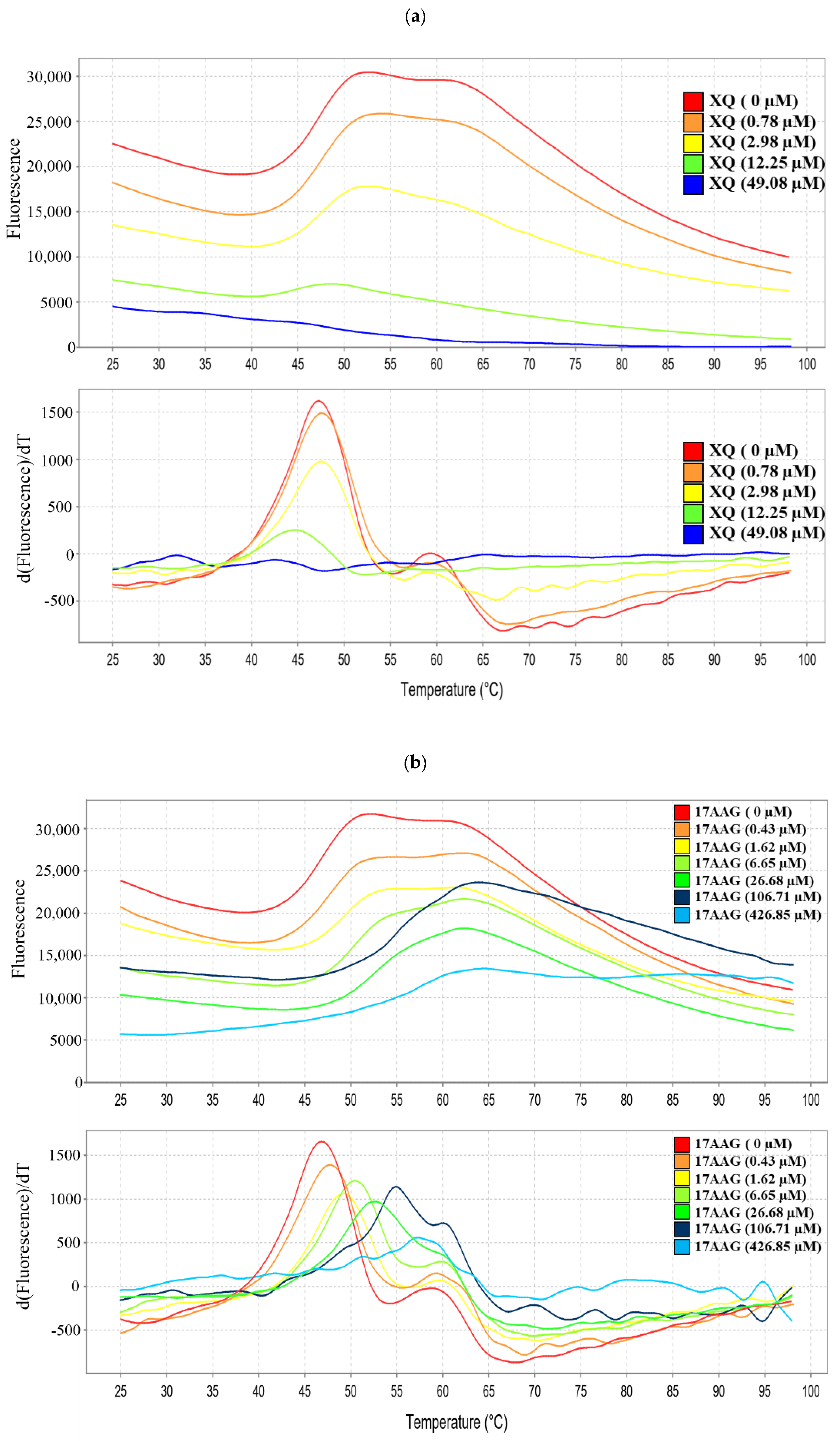
2.9. Interaction of Xestoquinone and HSP-90 Protein
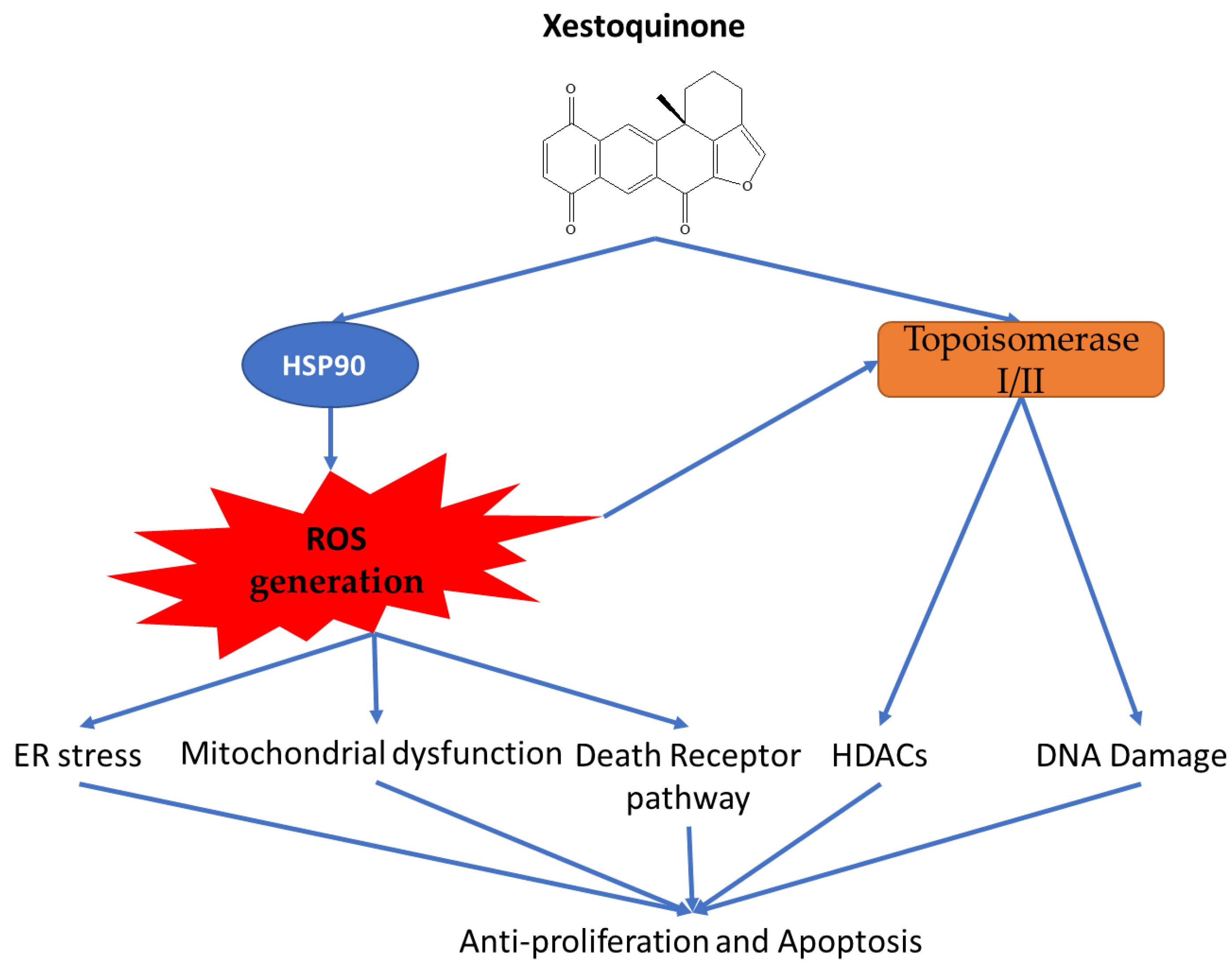
3. Discussion
4. Materials and Methods
4.1. Chemicals and Biological Materials
4.2. Preparation of Xestoquinone Stock Solution
4.3. Assay of Pan-Histone Deacetylase Activity
4.4. Annexin V/PI Apoptotic Assay
4.5. Determination of ROS Generation, Calcium Accumulation, and MMP Disruption
4.6. Western Blotting Analysis
4.7. Immunofluorescence Analysis
4.8. Human Leukemia Molt-4 Cells’ Xenograft Animal Model
4.9. Histopathological Evaluation
4.10. Molecular Docking Assay
4.11. Protein Thermal Shift Assay
4.12. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pal, A.; Chakravarty, A.K. (Eds.) Chapter 9—Genes conferring immunity against bacterial infections. In Genetics and Breeding for Disease Resistance of Livestock; Academic Press: Cambridge, UK, 2020; pp. 153–161. [Google Scholar]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.; Taguwa, S.; Frydman, J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 2012, 1823, 698–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, X.; Campos, L.; Mounier, C.; Cornillon, J.; Flandrin, P.; Le, Q.H.; Piselli, S.; Guyotat, D. Expression of heat-shock proteins is associated with major adverse prognostic factors in acute myeloid leukemia. Leuk. Res. 2005, 29, 1049–1058. [Google Scholar] [CrossRef]
- Hazza, S.; Azzat, A.; El Shora, O.; El-Bedwey, M. Study of the role of HSP90 in acute myeloid leukemia. Egypt J. Haematol. 2014, 39, 72–79. [Google Scholar] [CrossRef]
- Radujkovic, A.; Schad, M.; Topaly, J.; Veldwijk, M.R.; Laufs, S.; Schultheis, B.S.; Jauch, A.; Melo, J.V.; Fruehauf, S.; Zeller, W.J. Synergistic activity of imatinib and 17-AAG in imatinib-resistant CML cells overexpressing BCR-ABL-Inhibition of P-glycoprotein function by 17-AAG. Leukemia 2005, 19, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Koll, T.T.; Feis, S.S.; Wright, M.H.; Teniola, M.M.; Richardson, M.M.; Robles, A.I.; Bradsher, J.; Capala, J.; Varticovski, L. HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Mol. Cancer Ther. 2008, 7, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Rong, B.; Yang, S. Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: New discoveries and developments (Review). Int. J. Oncol. 2018, 52, 321–336. [Google Scholar] [CrossRef]
- Park, H.K.; Yoon, N.G.; Lee, J.E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Yuno, A.; Lee, M.J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Methods Mol. Biol. 2018, 1709, 423–441. [Google Scholar]
- Banerji, U. Heat shock protein 90 as a drug target: Some like it hot. Clin. Cancer Res. 2009, 15, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Takano, H.; Kohno, K.; Matsuo, K.; Matsuda, T.; Kuwano, M. DNA topoisomerase-targeting antitumor agents and drug resistance. Anticancer Drugs 1992, 3, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Mohamadi Farsani, F.; Ganjalikhany, M.R.; Dehbashi, M.; Naeini, M.M.; Vallian, S. Structural basis of DNA topoisomerase II-α (Top2-α) inhibition: A computational analysis of interactions between Top2-α and its inhibitors. Med. Chem. Res. 2016, 25, 1250–1259. [Google Scholar] [CrossRef]
- Nitiss, J.L.; Nitiss, K.C. Resistance to Topoisomerase-Targeting Agents. In Encyclopedia of Cancer, 2nd ed.; Bertino, J.R., Ed.; Academic Press: New York, NY, USA, 2002; pp. 129–141. [Google Scholar]
- Nicolson, G.L.; Conklin, K.A. Reversing mitochondrial dysfunction, fatigue and the adverse effects of chemotherapy of metastatic disease by molecular replacement therapy. Clin. Exp. Metastasis 2008, 25, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Y.; Luo, X.; Hu, Q. Oxidative resistance of leukemic stem cells and oxidative damage to hematopoietic stem cells under pro-oxidative therapy. Cell Death Dis. 2020, 11, 291. [Google Scholar] [CrossRef]
- Walsby, E.J.; Coles, S.J.; Knapper, S.; Burnett, A.K. The topoisomerase II inhibitor voreloxin causes cell cycle arrest and apoptosis in myeloid leukemia cells and acts in synergy with cytarabine. Haematologica 2011, 96, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felix, C.A. Leukemias related to treatment with DNA topoisomerase II inhibitors. Med. Pediatr. Oncol. 2001, 36, 525–535. [Google Scholar] [CrossRef]
- Barker, C.R.; Hamlett, J.; Pennington, S.R.; Burrows, F.; Lundgren, K.; Lough, R.; Watson, A.J.; Jenkins, J.R. The topoisomerase II-Hsp90 complex: A new chemotherapeutic target? Int. J. Cancer 2006, 118, 2685–2693. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Are free radicals involved in thiol-based redox signaling? Free Radic. Biol. Med. 2015, 80, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2017, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Pallepati, P.; Averill-Bates, D.A. Mild thermotolerance induced at 40 °C protects HeLa cells against activation of death receptor-mediated apoptosis by hydrogen peroxide. Free Radic. Biol. Med. 2011, 50, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.P.; Lee, M.G.; El-Shazly, M.; Juan, Y.S.; Wen, Z.H.; Du, Y.C.; Su, J.H.; Sung, P.J.; Chen, Y.C.; Yang, J.C.; et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, R.P.; Jablonksy, M.J.; Shadid, M.; Romaine, I.; Dunlap, N.; Anklin, C.; Graves, D.E.; Osheroff, N. Substituents on etoposide that interact with human topoisomerase IIalpha in the binary enzyme-drug complex: Contributions to etoposide binding and activity. Biochemistry 2008, 47, 4501–4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilstermann, A.M.; Bender, R.P.; Godfrey, M.; Choi, S.; Anklin, C.; Berkowitz, D.B.; Osheroff, N.; Graves, D.E. Topoisomerase II—Drug interaction domains: Identification of substituents on etoposide that interact with the enzyme. Biochemistry 2007, 46, 8217–8225. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.C.; Valkov, N.; Yang, W.M.; Gump, J.; Sullivan, D.; Seto, E. Histone deacetylase interacts directly with DNA topoisomerase II. Nat. Genet. 2000, 26, 349–353. [Google Scholar] [CrossRef]
- Shimamura, Y.; Noaki, R.; Kurokawa, A.; Utsumi, M.; Hirai, C.; Kan, T.; Masuda, S. Effect of (-)-Epigallocatechin Gallate on Activation of JAK/STAT Signaling Pathway by Staphylococcal Enterotoxin A. Toxins 2021, 13, 609. [Google Scholar] [CrossRef]
- Lambert, L.J.; Grotegut, S.; Celeridad, M.; Gosalia, P.; Backer, L.J.; Bobkov, A.A.; Salaniwal, S.; Chung, T.D.; Zeng, F.Y.; Pass, I.; et al. Development of a Robust High-Throughput Screening Platform for Inhibitors of the Striatal-Enriched Tyrosine Phosphatase (STEP). Int. J. Mol. Sci. 2021, 22, 4417. [Google Scholar] [CrossRef] [PubMed]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Sviben, D.; Bertosa, B.; Hlousek-Kasun, A.; Forcic, D.; Halassy, B.; Brgles, M. Investigation of the thermal shift assay and its power to predict protein and virus stabilizing conditions. J. Pharm. Biomed. Anal. 2018, 161, 73–82. [Google Scholar] [CrossRef]
- Yu, Q.; Jiang, Y.; Sun, Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm. Sin. B 2020, 10, 746–765. [Google Scholar] [CrossRef]
- Du, L.; Mahdi, F.; Datta, S.; Jekabsons, M.B.; Zhou, Y.D.; Nagle, D.G. Structures and mechanisms of antitumor agents: Xestoquinones uncouple cellular respiration and disrupt HIF signaling in human breast tumor cells. J. Nat. Prod. 2012, 75, 1553–1559. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Liu, Y.C.; El-Shazly, M.; Shih, S.P.; Du, Y.C.; Hsu, Y.M.; Lin, H.Y.; Chen, Y.C.; Wu, Y.C.; Yang, S.C.; et al. The Antioxidant from Ethanolic Extract of Rosa cymosa Fruits Activates Phosphatase and Tensin Homolog In Vitro and In Vivo: A New Insight on Its Antileukemic Effect. Int. J. Mol. Sci. 2019, 20, 1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.C.; Peng, B.R.; Hsu, K.C.; El-Shazly, M.; Shih, S.P.; Lin, T.E.; Kuo, F.W.; Chou, Y.C.; Lin, H.Y.; Lu, M.C. 13-Acetoxysarcocrassolide Exhibits Cytotoxic Activity Against Oral Cancer Cells Through the Interruption of the Keap1/Nrf2/p62/SQSTM1 Pathway: The Need to Move Beyond Classical Concepts. Mar. Drugs 2020, 18, 382. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, C.; Long, G.; Wolf, J.; Okerberg, C.; Herbert, R. Qualitative and quantitative analysis of nonneoplastic lesions in toxicology studies. Toxicol. Pathol. 2002, 30, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeadIT B. Available online: http://www.biosolveit.de/LeadIT (accessed on 12 January 2011).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skok, Z.; Zidar, N.; Kikelj, D.; Ilas, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2020, 63, 884–904. [Google Scholar] [CrossRef] [PubMed]
- El-Shafey, H.W.; Gomaa, R.M.; El-Messery, S.M.; Goda, F.E. Synthetic approaches, anticancer potential, HSP90 inhibition, multitarget evaluation, molecular modeling and apoptosis mechanistic study of thioquinazolinone skeleton: Promising antibreast cancer agent. Bioorg. Chem. 2020, 101, 103987. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Types | Cell Lines | XQ IC50 (µM) |
|---|---|---|
| Leukemia cancer cells | Molt-4 | 2.95 ± 0.21 |
| K562 | 6.22 ± 0.21 | |
| Lymphoma cancer cells | Sup-T1 | 8.58 ± 0.60 |
| U937 | 11.12 ± 0.19 | |
| Normal rat macrophage | NR8383 | >30 |
| Sample | Tm Values (°C) |
|---|---|
| HSP-90 | 47.33 ± 2.53 |
| HSP-90+17-AAG (1.25 μg/mL; 106.71 μM) | 54.88 ± 3.44 *** |
| HSP-90+17-AAG (5 μg/mL; 426.85 μM) | 55.76 ± 3.46 *** |
| HSP-90+XQ (1.25 μg/mL; 196.32 μM) | 84.25 ± 1.90 *** |
| HSP-90+XQ (5 μg/mL; 785.28 μM) | 84.95 ± 0.86 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.-C.; Lu, M.-C.; Hsu, K.-C.; El-Shazly, M.; Shih, S.-P.; Lien, S.-T.; Kuo, F.-W.; Yang, S.-C.; Chen, C.-L.; Yang, Y.-C.S.H. The Antileukemic Effect of Xestoquinone, A Marine-Derived Polycyclic Quinone-Type Metabolite, Is Mediated through ROS-Induced Inhibition of HSP-90. Molecules 2021, 26, 7037. https://doi.org/10.3390/molecules26227037
Wang K-C, Lu M-C, Hsu K-C, El-Shazly M, Shih S-P, Lien S-T, Kuo F-W, Yang S-C, Chen C-L, Yang Y-CSH. The Antileukemic Effect of Xestoquinone, A Marine-Derived Polycyclic Quinone-Type Metabolite, Is Mediated through ROS-Induced Inhibition of HSP-90. Molecules. 2021; 26(22):7037. https://doi.org/10.3390/molecules26227037
Chicago/Turabian StyleWang, Kuan-Chih, Mei-Chin Lu, Kai-Cheng Hsu, Mohamed El-Shazly, Shou-Ping Shih, Ssu-Ting Lien, Fu-Wen Kuo, Shyh-Chyun Yang, Chun-Lin Chen, and Yu-Chen S. H. Yang. 2021. "The Antileukemic Effect of Xestoquinone, A Marine-Derived Polycyclic Quinone-Type Metabolite, Is Mediated through ROS-Induced Inhibition of HSP-90" Molecules 26, no. 22: 7037. https://doi.org/10.3390/molecules26227037
APA StyleWang, K. -C., Lu, M. -C., Hsu, K. -C., El-Shazly, M., Shih, S. -P., Lien, S. -T., Kuo, F. -W., Yang, S. -C., Chen, C. -L., & Yang, Y. -C. S. H. (2021). The Antileukemic Effect of Xestoquinone, A Marine-Derived Polycyclic Quinone-Type Metabolite, Is Mediated through ROS-Induced Inhibition of HSP-90. Molecules, 26(22), 7037. https://doi.org/10.3390/molecules26227037