2.1. Structural and Optical Properties
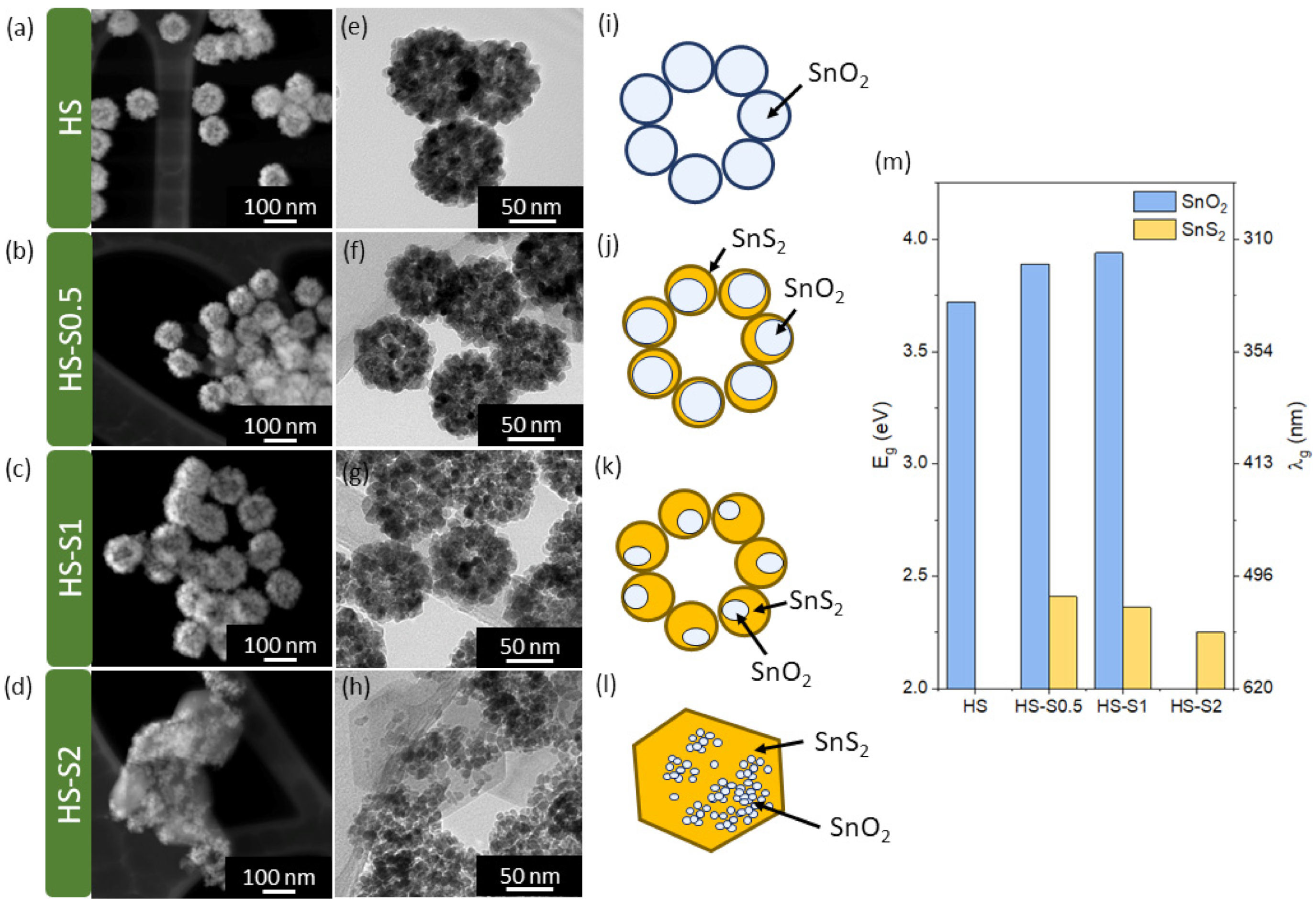
The synthesis parameters of the analyzed materials, as well as their (micro)structural, surface, and optical properties, are presented in
Table 1. The formation mechanism of SnO
2/SnS
2 heterostructures and in-depth characterization of their structure using X-ray diffraction (XRD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and high-resolution transmission electron microscopy (HRTEM) were reported in our previous work [
19]. Briefly, the SnO
2/SnS
2 heterostructures were obtained via chemical conversion of hollow-spherical raspberry-like SnO
2 nanomaterials (HS). In the syntheses, we used three different concentrations (
Table 1) of a sulfide precursor (thioacetamide, TAA). Depending on the applied TAA:HS molar ratio, the synthesized samples exhibited different structural properties. For TAA:HS = 0.5 and TAA:HS = 1 (samples labeled HS-S0.5 and HS-S1, respectively), multi-core@shell SnO
2@SnS
2 nanomaterials were obtained. The reported multi-core@shell structure can be described as multiple single-core@shells that form a raspberry-like shape (see
Figure 1j–k). The application of a higher TAA concentration resulted in an increased SnS
2 content in the samples. For HS-S2 (TAA:HS = 2), however, the core morphology underwent disintegration.
Transmission and scanning transmission electron microscopy (TEM and STEM) images of the samples are presented in
Figure 1a–h. The obtained results confirm that only in the case of HS-S2, the raspberry-like shape was completely disintegrated. It can be seen that this sample consists of larger grains surrounded by randomly distributed smaller elements. The measured grain size values of all samples are listed in
Table 1. Moreover, since the analyzed samples are potential candidates for photocatalysts, their optical properties were examined. Based on the measurements of total reflectance spectra, the absorption edge (λ
g) and bandgap values (E
g) were determined (
Table 1,
Figure 1m). As expected, the HS sample can absorb only UV light and its bandgap (E
g = 3.7 eV) corresponds to SnO
2 [
20]. All HS-S samples, in turn, exhibit light absorption in the visible range. Bandgap values of about 2.3 eV can be assigned to SnS
2 [
14]. With an increasing SnS
2 content, the absorption edge (λ
g) from this component shifts towards longer wavelengths. Samples that preserve the raspberry-like shape (HS-S0.5, HS-S1) are characterized by E
g values corresponding to both the SnO
2 and SnS
2 components. In the case of HS-S2, in turn, only one bandgap from SnS
2 was detected. This may indicate that the SnO
2 core is covered with a thick layer of sulfide.
2.2. Adsorptive Properties
Adsorption of pollutants on the catalyst surface is the first step of a photodecomposition process. For this reason, the overall efficiency of a photocatalyst is strictly related to its surface properties. Depending on the net surface charge in aqueous solutions, a photocatalyst would have a higher affinity towards positively or negatively charged molecules. Therefore, zeta potential measurements were carried out (
Supplementary Materials Figure S1) to characterize how the surface charge of the samples’ dispersions changes with pH. Based on the obtained results, all the samples characterize a negative surface charge over a wide pH range. It can therefore be assumed that they would more likely adsorb molecules with a net positive charge. The isoelectric point values (IEP) are listed in
Table 1. Compared to HS, the IEP of the SnO
2/SnS
2 composites shift towards the acidic region. The lowest IEP value was recorded for the HS-S2 sample (IEP = 4.24) with the highest SnS
2 content. Moreover, it is worth noting that HS has absolute zeta potential values (|ZP|) higher than 30 mV only in solutions with pH > 7.6. For SnO
2/SnS
2 heterostructures, in turn, such values were obtained in pH > 6.3 (HS-S0.5) and pH > 5.3 (HS-S1, HS-S2). Thus, the SnO
2/SnS
2 dispersions (HS-S0.5, HS-S1, and HS-S2) can be assumed to be stable in a wider pH range than pure SnO
2 (HS) [
13].
For a better understanding of the nanoparticle dispersions’ properties, hydrodynamic diameter (d
h) measurements were performed. This parameter allows characterizing the agglomeration tendency of nanopowders in aqueous solutions [
21]. Moreover, the analysis of d
h values makes it possible to indirectly evaluate the adsorption behavior of powders towards various molecules, such as organic dyes [
22]. Therefore, we conducted d
h measurements in deionized water (DIW) and aqueous solutions containing dyes with a net positive/negative charge. We chose rhodamine B (RhB) as a representative of cationic dyes and indigo carmine (IC) as anionic ones. The results are presented in
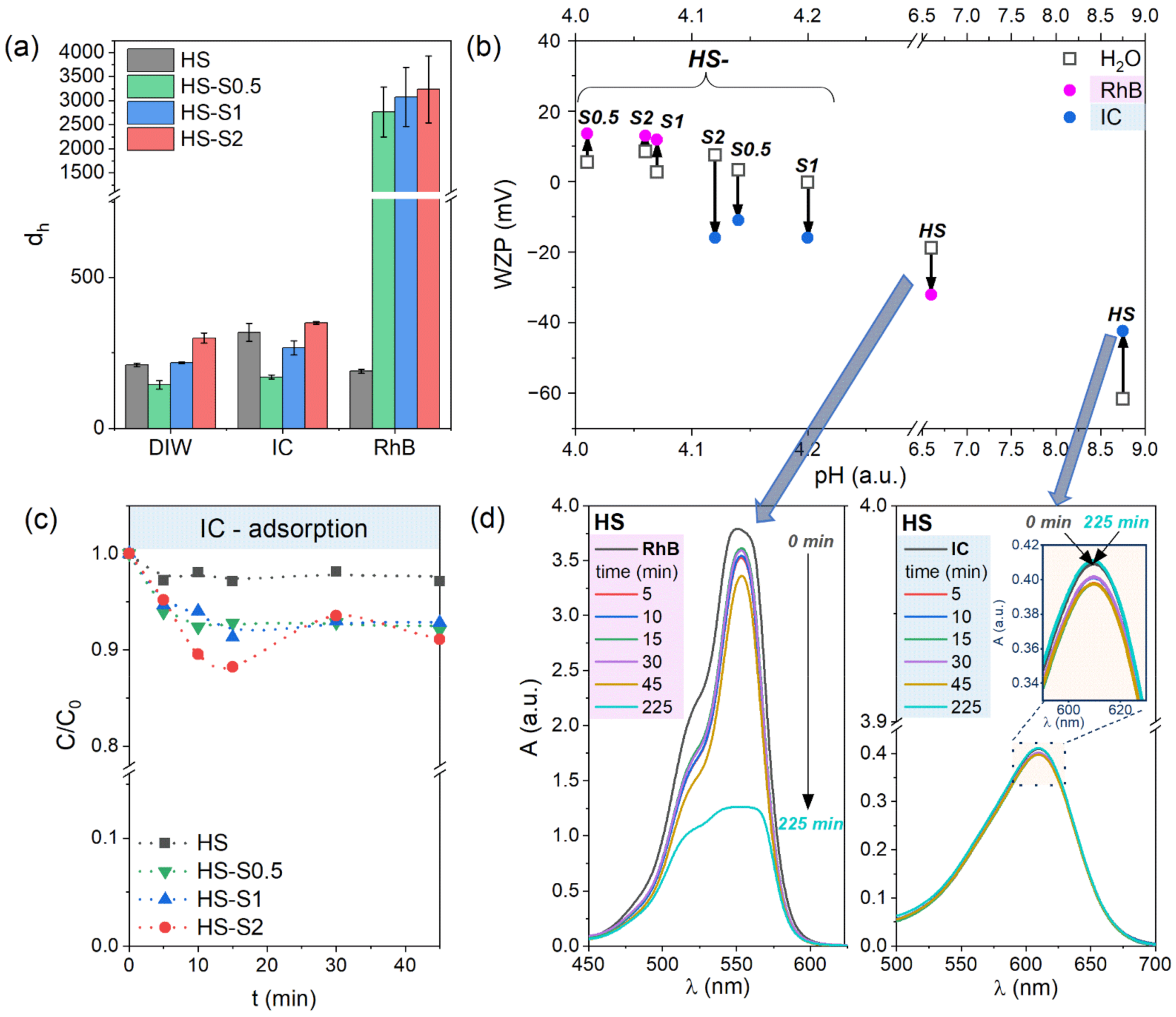
Figure 2a. The hydrodynamic diameter values obtained in DIW are also listed in
Table 1. It can be clearly seen that despite the similar grain size (
Table 1), the HS-S0.5 sample has a lower d
h than the HS in DIW. This may indicate that HS nanoparticles have a higher tendency to agglomerate. Consequently, their reactivity might be reduced [
21]. The obtained results are consistent with zeta potential measurements, which indicate that HS-S0.5 characterize higher |ZP| values. Both samples (HS and HS-S0.5) also show a similar tendency in anionic IC solutions. For HS-S1 and HS-S2, the d
h values are higher than in the case of HS despite the more stable suspensions of these compounds. Higher hydrodynamic diameter values may be connected with larger grain size and specific surface area of HS-S1 and HS-S2 (
Figure S2). The presented BET results revealed that with an increasing SnS
2 content in the samples, the specific surface area (S
BET) of the obtained structures is higher. Therefore, it could be assumed that the HS-S2 sample would adsorb the highest amount of dyes.
For all samples, the average hydrodynamic diameter increase in IC solutions compared to DIW (318, 170, 267, and 349 nm for HS, HS-S0.5, HS-S1, and HS-S2, respectively). This increase may indicate the adsorption of dye molecules on the powders’ surface. Starkly different results were obtained for cationic rhodamine B solutions. In the case of HS, the average hydrodynamic diameter decreased compared to DIW (189 and 210 nm, respectively). Thus, RhB adsorption is highly likely to have affected the breakdown of HS agglomerates into smaller groups. For all SnO
2/SnS
2 heterostructures, in turn, the measured d
h values were approximately 10 times higher than in DIW and IC. On the one hand, this may indicate the strong affinity of these samples for cationic dyes and therefore confirm the assumption based on zeta potential measurements. On the other hand, these higher values may be related to the agglomeration of nanoparticles induced by the adsorption of RhB [
22]. According to Talbot et al. [
22], such agglomeration can affect the spatial distribution of dye molecules and, consequently, have an impact on adsorption equilibrium. This effect was also observed during our experiment. For each measurement of the SnO
2/SnS
2 samples in RhB, the obtained d
h values differed significantly from each other (see error bars in
Figure 2a). Therefore, it can be assumed that the RhB adsorption equilibrium can be reached only in the case of the HS sample.
Wall zeta potential (WZP) is a parameter that can be used to study the adsorption kinetics of various soluble components [
23]. When particles adsorb on the walls of the measuring vessel, its surface charge (wall zeta potential) changes. This measurement is very sensitive to any changes in the system, including the presence of additional ions or a change in the solution pH. Any modification of the analyzed material changes its ability to deposit. As a result of the dye adsorption on the adsorbent/photocatalyst’s surface, the target group of its application can be determined.
Figure 2b presents how the WZP of the suspensions changes in RhB/IC compared to aqueous solutions of the same pH. For IC, it can be seen that the wall zeta potential of HS shifts towards higher values, while for HS-S composites (i.e., HS-S0.5, HS-S1, and HS-S2), it decreases. The opposite tendency was observed in the case of RhB solutions. Therefore, it can be assumed that HS characterizes adsorption behavior different from that of its heterostructures (HS-S). For the RhB solution, the WZP of HS shifts towards lower values, indicating that the dispersion is more stable. This is consistent with d
h measurements, which revealed that RhB adsorption caused the breakdown of agglomerates and therefore stabilized the suspension. In the case of the HS-S samples, in turn, WZP slightly shifted towards higher values. This suggests that the analyzed suspensions are not stable and have a tendency to agglomerate, which also confirmed the d
h results. It is also worth noting that the pH values of the suspensions in RhB suggest that HS-S powders have a net positive charge (see
Figure S1), whereas HS is negatively charged. This implicates that HS-S samples repulse the molecules of cationic RhB, while HS attracts them. For the IC solution, the WZP of HS increased, indicating lower stability of the suspension. The reverse nature of changes for HS-S, in turn, suggests that their suspensions in IC are more stable. This is also consistent with zeta potential measurements. The HS-S suspensions in IC are positively charged, while HS characterize a net negative charge (
Figure S1). Thus, HS-S attracts anionic IC molecules and HS repulses them. Based on the obtained results, it can therefore be assumed that the HS sample would characterize better adsorption properties towards RhB, while HS-S composites towards IC.
The adsorption properties of the powders were further analyzed by performing adsorption tests. Briefly, the suspension containing the dye solution and dispersed powder was continuously stirred in the dark at room temperature and under atmospheric pressure. At specific time intervals, the suspension was collected, and the powder was separated from the dye solution. Relative concentrations (C/C
0) were determined from changes in absorbance intensity (A/A
0) of powder-free solutions. The experimental setup is presented in
Figure S3 (for adsorption tests, the light was turned off).
Figure 2c presents the adsorption kinetics of IC on the studied samples. The obtained curves suggest that adsorption equilibrium was reached after 45 min of stirring in the dark for HS, HS-S0.5, and HS-S1. The amount of adsorbed dye was 3% for HS, while for HS-S0.5 and HS-S1 it was 7%. In the case of HS-S2, the process did not stabilize. Nevertheless, extending the experiment duration to approximately 4 h revealed a different adsorption behavior of the powders. For HS, the measured absorbance of the IC sample collected after 225 min (
Figure 2d) suggests that the initially adsorbed amount of dye was almost completely desorbed. The results for HS-S0.5, HS-S1, and HS-S2 are presented in
Figure S4. HS-S0.5 showed behavior similar to that of HS, however, the remaining amount of adsorbed dye was 4%. Only in the case of HS-S1, the absorbance of the sample did not change compared to that obtained after 45 min, indicating that the adsorption equilibrium was reached. The measured values for HS-S2 suggest that even after nearly 4 h the adsorption-desorption process did not stabilize. In the case of this powder, the amount of adsorbed IC was 19, 12, and 16% after 105, 165, and 225 min, respectively. The amounts of the adsorbed IC dye measured after different periods of time are presented in
Figure S5a. It can be seen that with an increasing SnS
2 content in the samples, they adsorbed a higher amount of IC after approximately 4 h of the experiment. This is consistent with the assumption based on the BET results. Analysis of the HS behavior towards RhB (
Figure 2d), in turn, revealed that it adsorbed 67% of the dye after 225 min of stirring in the dark. Thus, the obtained results confirm the assumption based on WZP measurements regarding the better adsorption properties of HS and HS-S towards RhB and IC, respectively. Nevertheless, only for HS-S1, the IC adsorption-desorption process stabilized. This indicates that in addition to the materials’ specific surface area, their (micro)structural properties also have an influence on the adsorption behavior.
Since the adsorption equilibrium was not reached for HS-S2, unlike for HS-S1, we decided to investigate the morphology of these samples after the IC adsorption process (
Figure S6). The presented SEM images reveal that the microstructure of HS-S2 underwent disintegration, while the hollow-spherical raspberry-like shape of HS-S1 remained unchanged. Therefore, it can be assumed that for the HS-S1 sample, the morphology of multi-core@shell SnO
2@SnS
2, as well as its surface properties (ZP and WZP) allow achieving the best adsorption properties towards IC.
2.3. Photocatalytic Properties
The photocatalytic properties of the samples were investigated by performing dye decomposition tests under visible light illumination (see the experimental setup in
Figure S3). Before illumination, the suspension containing the dye solution and the powder photocatalyst was stirred in the dark for 30 min. The samples were collected and analyzed in the same manner as for the dye adsorption tests. To reliably assess the photocatalytic performance of a catalyst, it is crucial to choose an appropriate dye for the tests. Based on the analysis of the adsorptive properties, we expected that HS-S composites would not decompose rhodamine B (RhB). Preliminary photocatalytic tests confirmed this assumption (
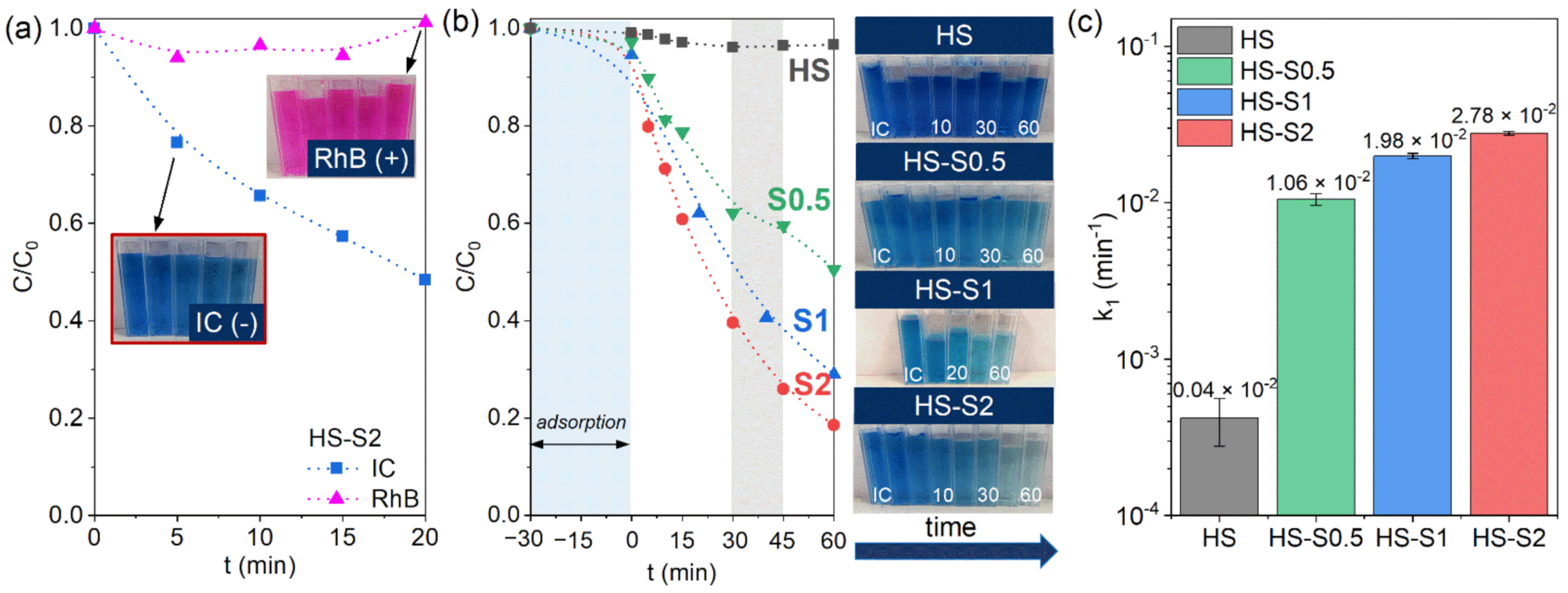
Figure 3a). After 20 min of illumination, visible light-active HS-S2 decomposed approximately 50% of indigo carmine (IC), while the RhB concentration remained almost unchanged. This is consistent with the ZP, WZP, and d
h measurements, which revealed that the HS-S particles repulse RhB molecules and the suspensions are not stable. Therefore, the photocatalytic process cannot occur without effective adsorption. Based on the obtained results, IC was chosen for further photocatalytic tests.
Figure 3b presents the IC decomposition kinetics obtained under visible light illumination using HS, HS-S0.5, HS-S1, and HS-S2 as photocatalysts. The results reveal that the analyzed powders decomposed (and/or adsorbed) 3, 49, 71, and 81% of IC after 60 min of illumination, respectively. In the case of HS, its wide bandgap value (3.7 eV) does not allow it to decompose dye under visible light. Sensitization of SnO
2 with visible light-active SnS
2 resulted in a considerable enhancement of photocatalytic activity. In the case of HS-S0.5, it can be seen that the nature of the IC concentration changes differs from HS-S1 and HS-S2. For instance, between 30 min and 45 min of the process, only 3% of the dye was decomposed. Such behavior may be the result of IC desorption from the HS-S0.5 surface, which was confirmed by adsorption tests (
Figure S4). It is also worth noting that despite the much higher SnS
2 content in HS-S2 (32.9 wt.%) than in HS-S1 (11.6 wt.%), there is a slight difference in photocatalytic efficiency (10%) of the samples.
The investigation of the IC decomposition process pointed out that it follows pseudo-first-order kinetics according to the Langmuir-Hinshelwood model [
24]. Therefore, the pseudo-first-order rate constants (k
1) were determined as a slope of the linear plot of ln(C
0/C) vs. irradiation time (
Figure S5b). The calculated k
1 values are presented in
Figure 3c. It can be clearly seen that even applying a low SnS
2 content (2.6 wt.% for HS-S0.5) considerably increases the photocatalytic efficiency of the systems. The highest k
1 value was obtained for HS-S2 (2.78 × 10
−2 min
−1). However, in the case of this sample, the adsorption tests (
Figure 2c,
Figures S4 and S5a) revealed that this process did not stabilize over time. Therefore, the observed photocatalytic efficiency is a result of both adsorption and photocatalysis.
The effect of solution pH on the photocatalytic performance of HS-S2 was also examined, and the results are presented in
Figure S7. The pH of the IC/HS-S2 solution without adjustment was 4. It can be seen that shifting pH towards the acidic region resulted in the deterioration of photocatalytic properties, while increasing pH to 5 led to better efficiency. A further increase in the pH solution, in turn, resulted in a reduced amount of decomposed dye. Despite a higher positive net charge of HS-S2 at pH = 3 (
Figure S1), the increased concentration of H
+ ions in the solution could cause anionic IC molecules to stay in the diffusion layer and thus hinder charge transfer from HS-S2 to IC [
4]. Therefore, the measured photocatalytic efficiency was reduced. On the other hand, despite a net negative charge of HS-S2 at pH = 5 (
Figure S1), the OH
− ions might push anionic IC molecules into the Stern layer and improve the charge transfer [
4]. Therefore, the obtained efficiency was higher. A more negative net charge of HS-S2 at pH = 6 could increase the electrostatic repulsion of IC molecules and, thus, hinder the photocatalytic performance. To eliminate the influence of additional H
+ and OH
− ions in the solution, further tests were performed without pH adjustment.
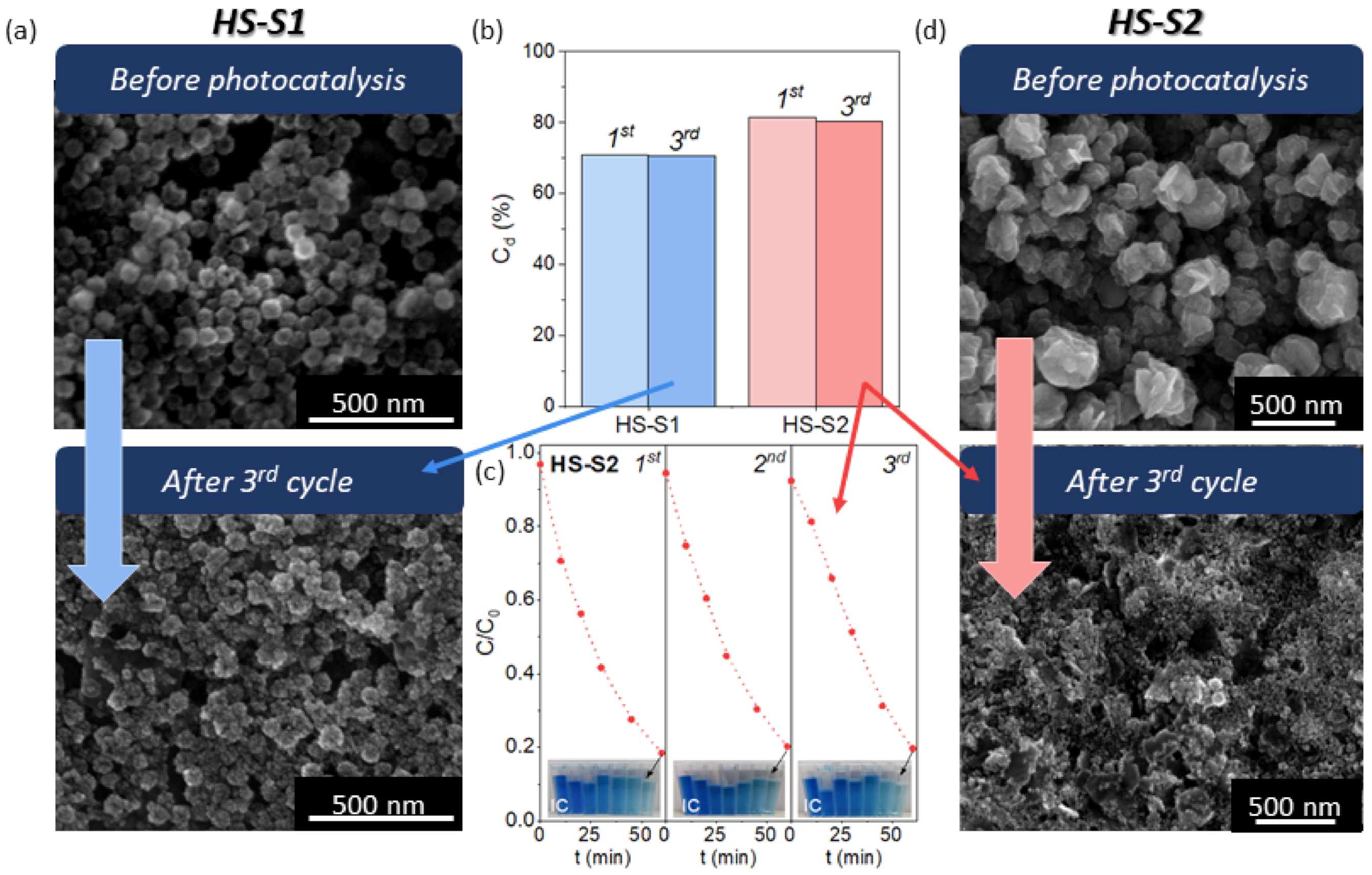
To assess the stability of the photocatalysts, we performed cyclic photodecomposition tests of IC using the samples that showed the best photocatalytic performance (HS-S2, HS-S1). Briefly, after each cycle, the powder was collected and reused for the next one. The obtained photodecomposition kinetics and efficiencies (C
d) after the first and third cycles are presented in
Figure 4. For both samples, the measured photocatalytic efficiency only slightly decreased after the third run. The decrease in C
d value was minimally higher for HS-S2. Based on these measurements, it might seem that both catalysts are stable. Nevertheless, the results of cyclic photodecomposition experiments are not sufficient to evaluate photostability [
18]. According to Chen et al. [
18], various factors may affect the photocatalytic efficiency, such as adsorption of intermediates and surface changes. Therefore, we analyzed the morphology of the samples after performing cyclic tests (
Figure 4). It was found that the microstructure of HS-S2 was completely disintegrated, whereas the shape of HS-S1 was preserved. The SEM images of HS-S1 collected after the first and second cycles are presented in
Figure S6. It can be seen that after each cycle the raspberry-like morphology of multi-core@shell SnO
2@SnS
2 remained unchanged. The obtained SEM micrographs are consistent with those received after adsorption tests (
Figure S6). They revealed that even IC adsorption (without photocatalysis) affects HS-S2 morphology. The microstructural disintegration of this sample might result in more sites available for dye adsorption. Therefore, despite the photocatalyst degradation, the measured efficiency after each cycle may appear unchanged. The BET results (
Figure S2) revealed that the HS-S2 sample collected after the first IC photodecomposition cycle (labeled HS-S2-1st) characterized a slightly lower specific surface area than before conducting the cyclic experiments. However, as it can be seen from the SEM images (
Figure 4 and
Figure S6), the sample after performing the adsorption/photocatalytic tests consisted of a very small elements of about 30 nm in size. Therefore, as a result of high surface energy, they may have a strong tendency to agglomerate. Thus, the measured S
BET may in fact be the surface area of agglomerates and, consequently, it may be understated.
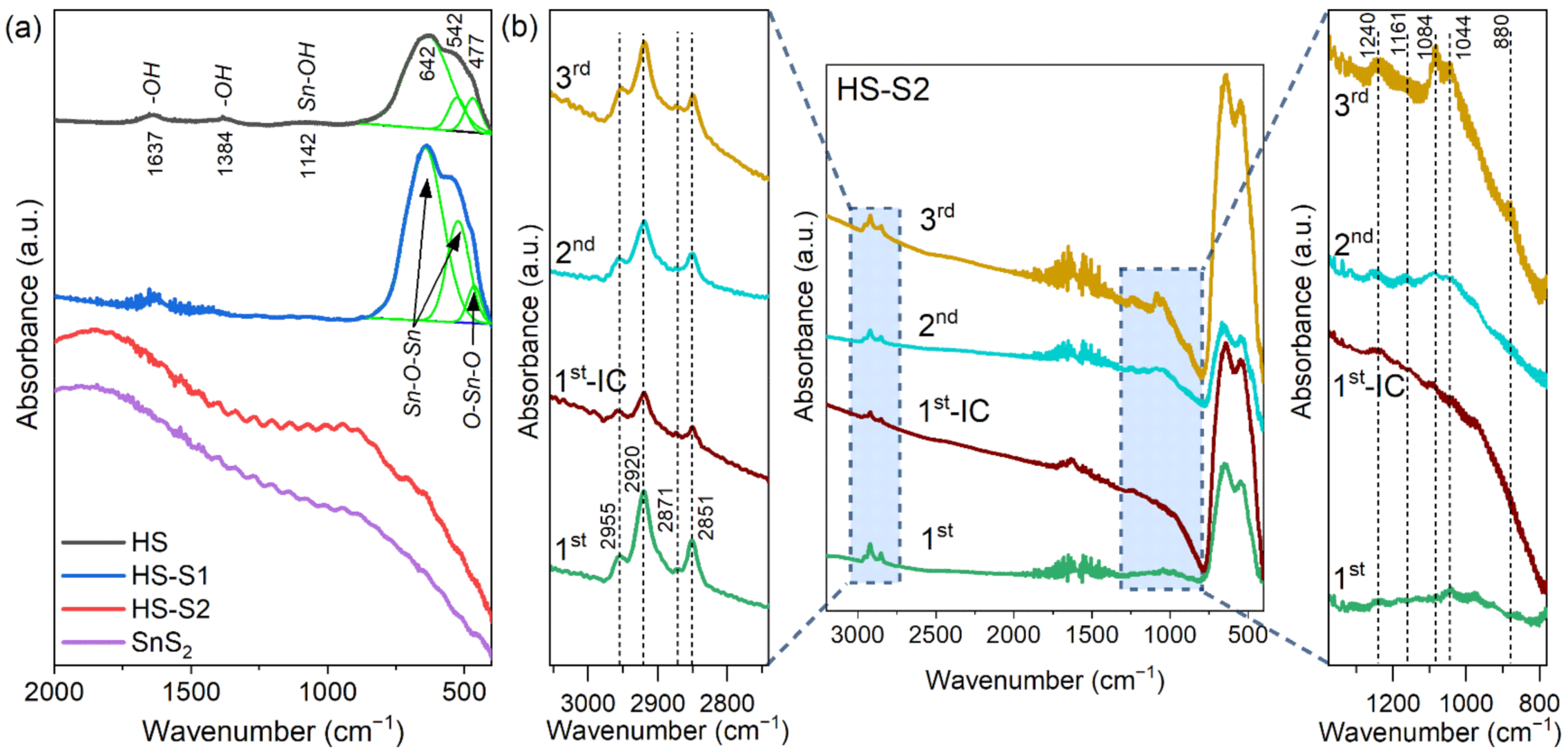
To better understand the changes in the surface state of the HS-S2 sample, FTIR spectroscopy was applied.
Figure 5a presents the results obtained for the as-synthesized HS, HS-S1, and HS-S2 samples, which were compared with pure SnS
2. The FTIR spectra of HS-S2 after each photodecomposition cycle are shown in
Figure 5b.
In the case of HS, the characteristic vibration bands for SnO
2 at ca. 542 cm
−1 and 642 cm
−1 are visible (
Figure 5a). These bands are typical for the Sn-O-Sn terminal oxygen vibrations mode. Moreover, the band at ca. 477 cm
−1 can be attributed to the O–Sn–O bridge functional group of SnO
2. The presence of additional absorption bands at 1384 and 1637 cm
−1 is related to hydroxyl groups of molecular water adsorbed on the sample’s surface. The peak at ca. 1142 cm
−1, in turn, can be considered as the Sn–OH stretching mode [
25,
26,
27].
With an increasing SnS
2 content in the samples, the vibration modes of tin dioxide are disappearing (
Figure 5a). The recorded spectrum for HS-S2 is similar to that of pure SnS
2. Therefore, it can be concluded that sulfide covers the SnO
2 core with a thick layer, which was also confirmed by optical measurements (
Figure 1m).
Figure 5b presents the results obtained for the HS-S2 sample collected after each photodecomposition cycle. Interestingly, after performing the first cycle (sample labeled 1st), the FTIR spectrum of HS-S2 no longer resembled that obtained for SnS
2. This confirms the SEM observations, which revealed the disintegration of the HS-S2 microstructure. Consequently, the characteristic bands for SnO
2 could be detected.
Furthermore, it should be noted that after each cycle the sample was rinsed with water/ethanol mixture (50/50 v/v) to remove unbound dye groups and dried at 25 °C under vacuum. For comparison purposes, one sample was dried immediately after collection without washing (the sample labeled 1st-IC). In each case, additional bands indicating the presence of residual dye were observed. The absorbance intensity of these bands is increasing after each cycle, which is the result of dye chemical bonding to the photocatalyst.
In the range of 800–1400 cm
−1 are present bands that can be attributed to N–O (880 cm
−1), S–O (1044 cm
−1), C–O (1084 cm
−1), and OC–O=H vibrations (1161 and 1240 cm
−1 (δ
O–H + ν
C–O)) [
28,
29]. These bands are characteristic of indigo carmine (IC). The vibration modes from 2800 to 3000 cm
−1 can be assigned to the asymmetric (2851, 2871 cm
−1) and symmetric (2920, 2955 cm
−1) C–H stretching in CH
3 and CH
2 chains [
28,
30]. It is also worth noting that the 1st-IC sample shows some degree of fluorescence, which is characteristic of organic compounds (background elevation). Therefore, this provides evidence of the chemical bonding of the dye decomposition residues to the photocatalyst.
In addition to the (micro)structural and surface analysis of the reused powders, we collected the IC solution after performing the first photodecomposition cycle. The sample (labeled IC-HS-S2-1st) was analyzed using inductively coupled plasma optical emission spectroscopy (ICP-OES) to determine the presence of tin ions in the solution. The obtained results confirmed the degradation of HS-S2 during the photocatalysis. The measured Sn ions concentration for IC-HS-S2-1st was 0.2 mg/L (1.7 × 10−6 M), while for the reference sample containing IC solution before photocatalysis it was below the detection limit of the method (<0.001 mg/L). Based on the obtained results, it can be concluded that during photocatalysis the HS-S2 sample undergoes morphological and chemical changes in composition. The residues of the reaction products chemically bind to the surface of the grains, changing their properties. Moreover, the metal-ions from HS-S2 are leached into the IC solution. Consequently, the material changes its microstructure. This, in the case of its use as a photocatalytic material, is not a desirable phenomenon.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}