4.3.5. General Procedure for the Reductive Amination of the Substituted Diphenyl Ether Aniline with the Substituted 2-Acetamide Benzaldehyde
To a solution of the diphenyl ether aniline (1.5 eq.) and aldehyde (1 eq.) in DCE (2 mL/1 mmol), acetic acid (3 eq.) and sodium tri-acetoxyborohydride (2.5 eq.) were added. The resulting reaction mixture was stirred at r.t. for 2–18 h. NaHCO3 was then added and repeatedly extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography to afford the desired compound.
2-Amino-benzaldehyde
Using the general procedure for the reduction of the 2-nitrobenzylalcohol, the desired compound was isolated as a yellow oil, 794 mg, 99% yield. R.f. 0.35 (DCM), LCMS: tr = 3.04 min (50% to 95% MeOH in water at 0.5 mL/min to 1.0 mL/min over 5 min), m/z M + H 122.15, 1H NMR (CDCl3, 270 MHz,): δ 6.12 (2H, s, NH2), 6.63 (1H, d, J = 8.4 HZ, ArH), 6.70–6.76 (1H, m, ArH), 7.26–7.32 (1H, m, ArH), 7.46 (1H, dd, J = 1.5, 7.7 Hz, ArH), 9.85 (1H, s, CHO).
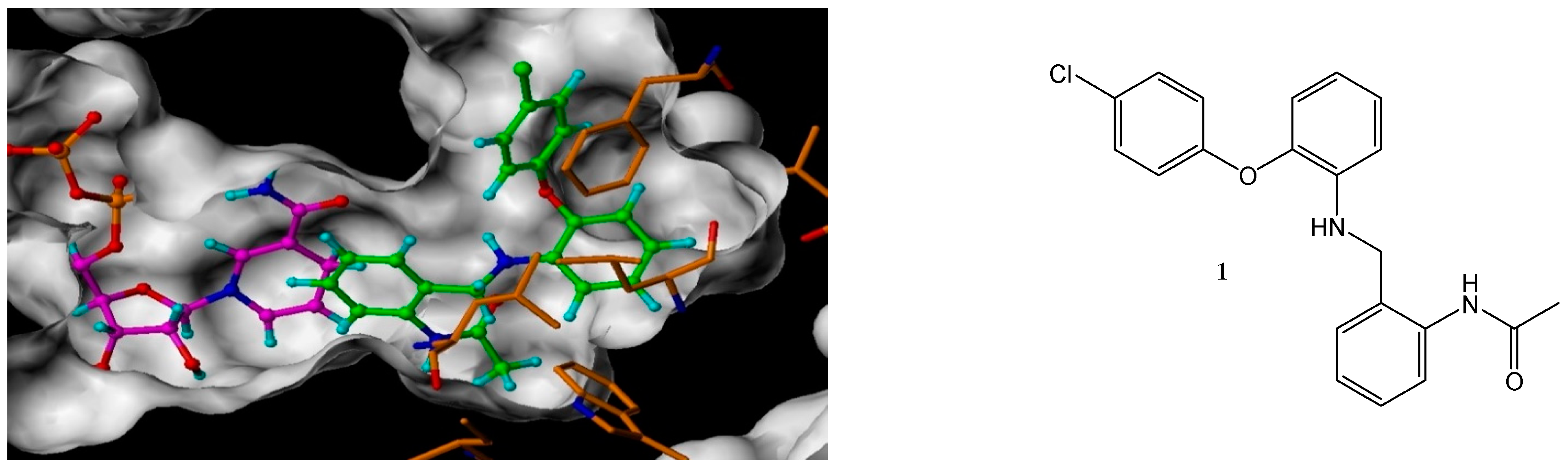
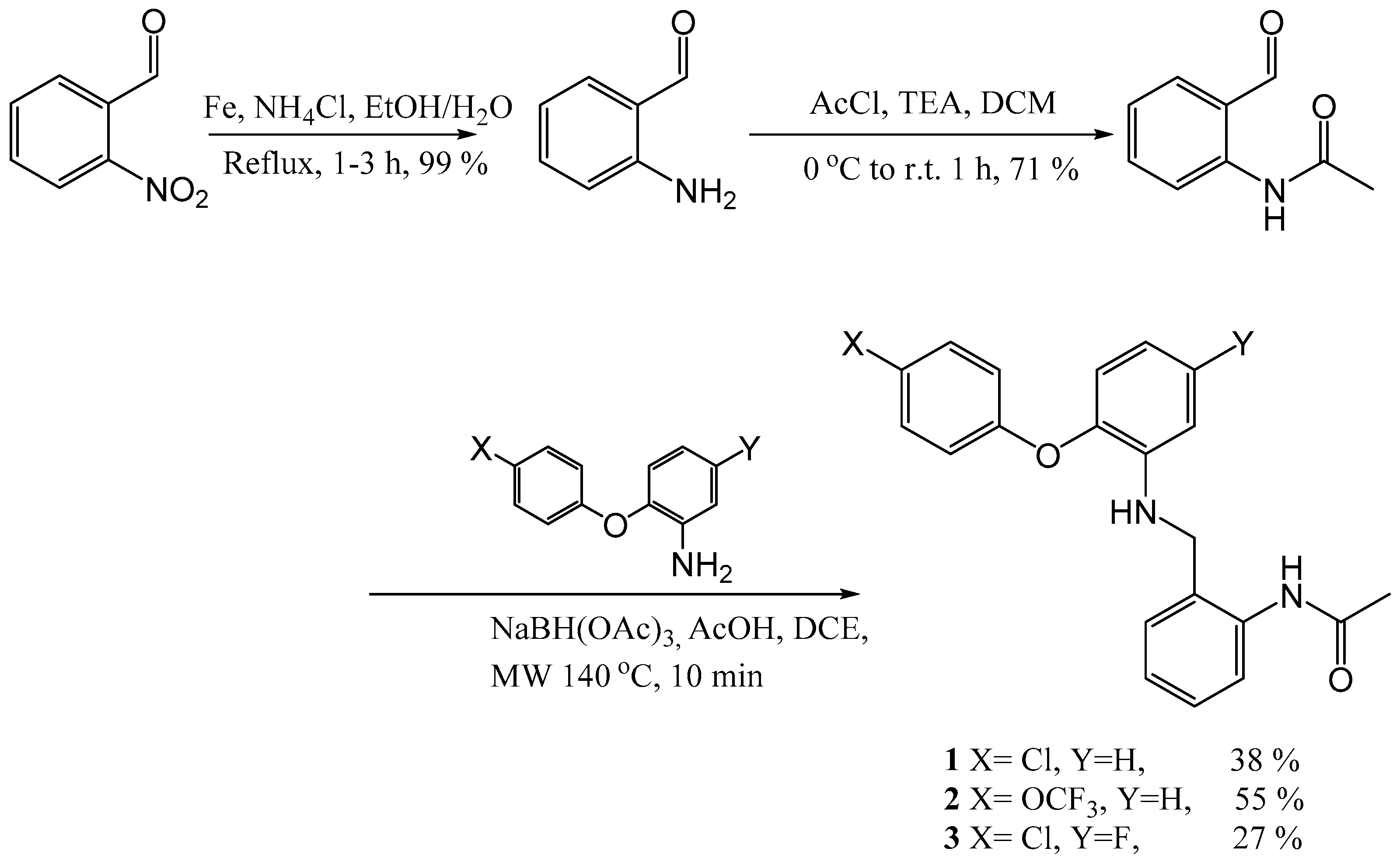
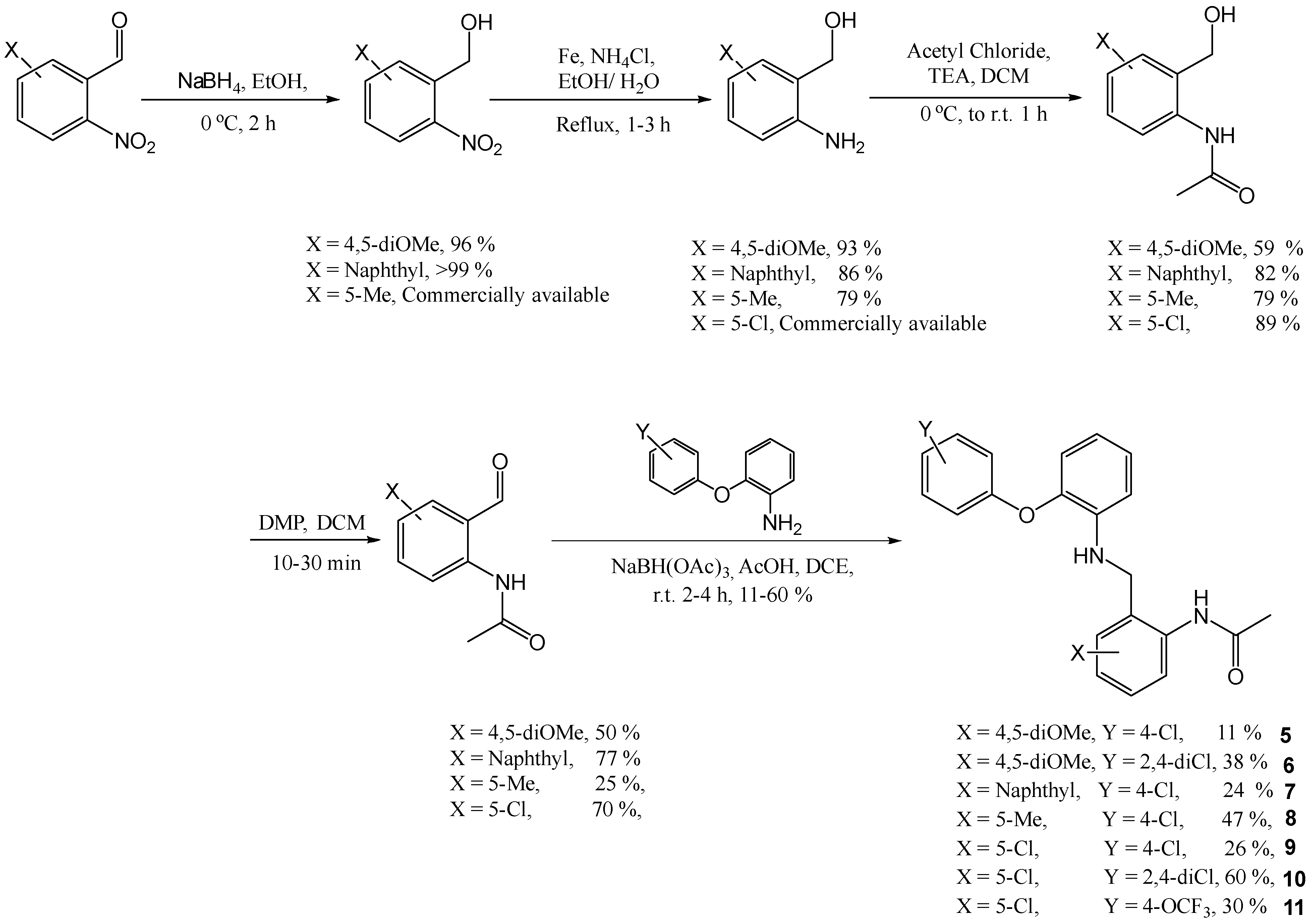
N-(2-([2-(4-Chloro-phenoxy)-phenylamino]-methyl)-phenyl)-acetamide (1)
To a solution of 2-(4-chloro-phenoxy)-phenylamine (0.128 g, 0.58 mmol) and N-(2-formyl-phenyl)-acetamide 12 (0.19 g, 1.16 mmol) in DCE (2.6 mL), acetic acid (0.25 mL) and sodium tri-acetoxy borohydride (0.31 g, 1.45 mmol) were added. The resulting reaction mixture was heated in a CEM microwave for 10 min at 140 °C. NaHCO3 was added and the mixture was repeatedly extracted with EtOAc. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography (0–100% DCM in hexane) to afford the title compound as a white solid, 80 mg, 38% yield. R.f. 0.33 (DCM), m.p. 194–196 °C, LCMS: tr = 1.36 min (95% MeOH in H2O), m/z M - H 365.4, HPLC: tr = 5.1 min (90% acetonitrile in H2O, 0.5 mL/min), 98%, 1H NMR (DMSO, 400 MHz): δ 2.02 (3H, s, CH3), 4.27 (2H, d, J = 5.6 Hz, CH2), 5.95 (1H, s, NH), 6.46 (1H, d, J = 8.4 Hz, ArH), 6.55 (1H, td, J = 0.8, 7.6 Hz, ArH), 6.84 (1H, dd, J = 1.6, 8.0, ArH), 6.89–6.95 (3H, m, ArH), 7.07–7.11 (1H, m, ArH), 7.16–7.22 (2H, m, ArH), 7.35–7.41 (3H, m, ArH), 9.47 (1H, br.s, NHCO). 13C NMR (DMSO, 101 MHz): 23.2 (CH3), 42.6 (CH2), 111.7, 116.0, 118.3, 120.2, 125.2, 125.3, 125.6 (ArCH), 126.0 (ArC), 126.7, 126.9, 129.6 (ArCH), 133.5, 135.8, 140.5, 141.6, 156.7 (ArC), 168.5 (CO). HRMS: Calcd. for C21H19ClN2O2 (M + H)+ 367.1208, found (M + H)+ 367.1204. Anal. Calcd. for C21H19ClN2O2: C 68.76, H 5.22, N 7.64%. Found: C 69.0, H 5.28, N 7.52%.
N-(2-([2-(4-Trifluoromethoxy-phenoxy)-phenylamino]-methyl)-phenyl)-acetamide (2)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a white solid, 155 mg, 55% yield. R.f. 0.4 (1:1, EtOAc: Hexane), m.p. 98–100 °C, LCMS: tr = 1.07 min (95% MeOH in H2O), m/z M - H 415.09, HPLC: tr = 2.22 min (90% acetonitrile in H2O), 99%, 1H NMR (CDCl3, 270 MHz): δ 1.94 (3H, s, CH3), 4.3 (3H, s, CH2 and NH), 6.79–6.96 (5H, m, ArH), 7.06–7.17 (3H, m, ArH), 7.25–7.34 (3H, m, ArH), 8.01 (1H, d, J = 8.15, ArH), 8.54 (1H, br.s, NHCO). 13C NMR (CDCl3, 68 MHz): δ 24.5 (CH3), 47.8 (CH2), 113.8, 118.1, 119.6, 119.7 (ArCH), 122.5 (ArC), 122.8, 124.6, 125.7 (ArCH), 127.6 (ArC), 128.9, 129.6 (ArCH), 137.5, 139.9, 143.9, 144.4 (ArC), 155.9 (OCF3), 168.5 (CO). 19F NMR (CDCl3, 376 MHz): δ - 58.29 (OCF3). HRMS: Calcd. for C22H19F3N2O3 (M + Na)+ 439.1240, found (M + Na)+ 439.1240. Anal. Calcd. for C22H19F3N2O3: C 63.46, H 4.60 N 6.73%. Found: C 63.5, H 4.62, N 7.0%.
N-(2-([2-(4-Chloro-phenoxy)-5′-fluoro-phenylamino]-methyl)-phenyl)-acetamide (3)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a white solid, 68 mg, 27% yield. R.f. 0.4 (1:1, EtOAc: Hexane), m.p. 178–180 °C, LCMS: tr = 0.98 min (95% MeOH in H2O), m/z M-H 383.28, HPLC: tr = 2.87 min (90% acetonitrile in H2O), 99%, 1H NMR (CDCl3, 270 MHz): δ 2.03 (3H, s, CH3), 4.23 (2H, d, J = 5.0 Hz, CH2), 4.42 (1H, t, J = 4.9 Hz, NH), 6.41–6.58 (2H, m, ArH), 6.80–6.88 (3H, m, ArH), 7.10 (1H, t, J = 7.2 Hz, ArH), 7.22–7.32 (4H, m, ArH), 7.86 (1H, d, J = 7.9 Hz, ArH), 8.19 (1H, br.s, NHCO). 13C NMR (CDCl3, 68 MHz): δ 24.3 (CH3), 46.8 (CH2), 100.7 (d, J = 28.1 Hz, ArH), 104.6 (d, J = 23.7 Hz, ArCH), 118.0 (ArCH), 120.8 (d, J = 10.0 Hz, ArCH), 123.6 (ArCH), 128.1 (d, J = 11.8 Hz, ArC), 128.9, 129.4, 129.9 (ArCH), 136.9, 139.2 (ArC), 141.3 (d, J = 10.6 Hz, ArC), 156.3, 158.8, 162.3 (ArC), 168.7 (CO). 19F NMR (CDCl3, 376 MHz): δ -115.43–115.57 (m, ArF). HRMS: Calcd. for C21H18ClFN2O2 (M + H)+ 383.0968, found (M + H)+ 383.0965.
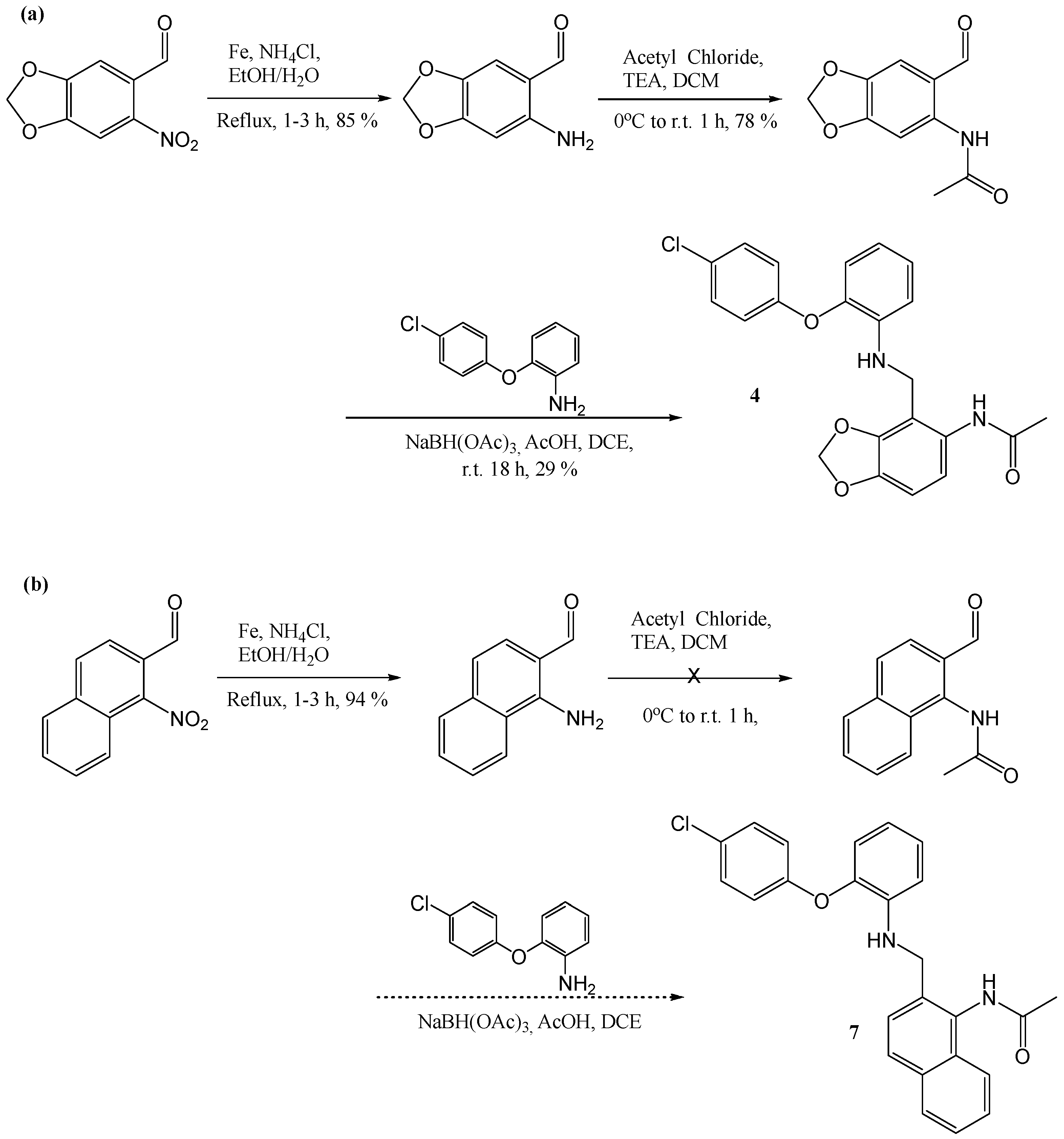
6-Amino-benzo[1,3]dioxole-5-carbaldehyde
Using the general procedure for the reduction of substituted 2-nitrobenzylalcohol, the desired compound was obtained as a brown solid, 360 mg, 85% yield. R.f. 0.67 (DCM), 1H NMR (CDCl3, 270 MHz,): δ 5.90 (2H, s, CH2), 6.11 (1H, s, ArH), 6.29 (2H, br.s, NH), 6.79 (1H, s, ArH), 9.57 (1H, s, CHO).
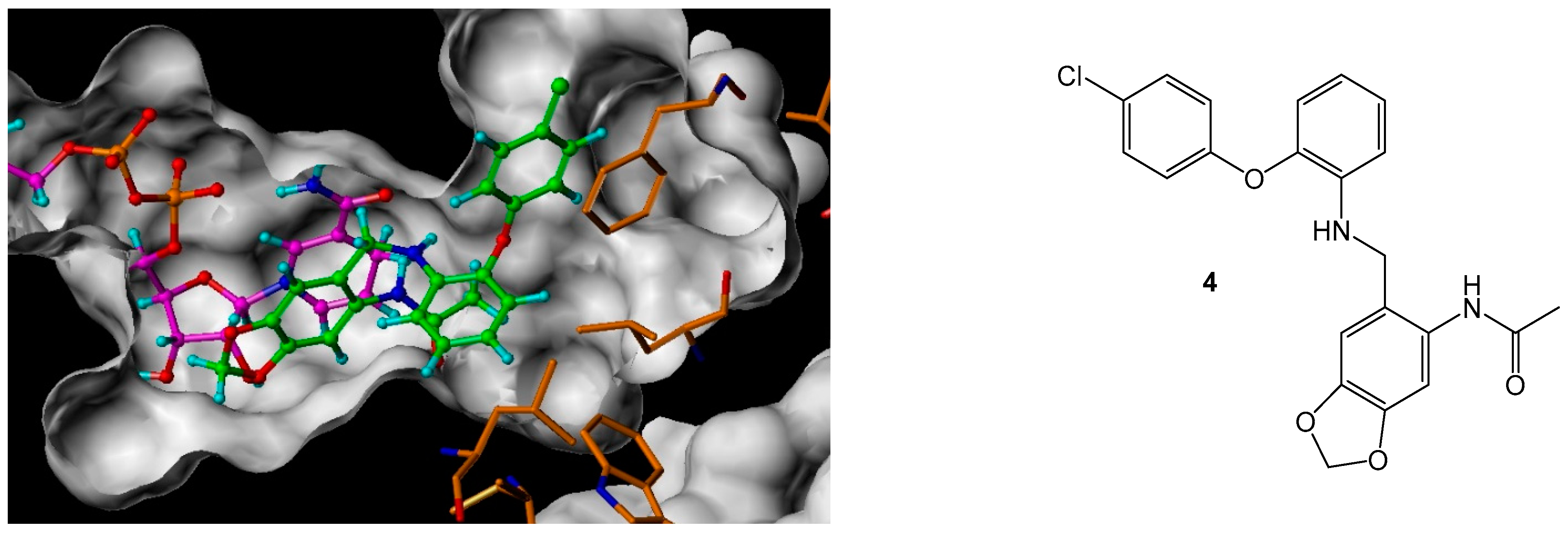
N-(6-[2-(4-Chloro-phenoxy)-phenylamino]-methyl-benzo[1,3]dioxol-5-yl)-acetamide (4)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a light cream solid, 540 mg, 29% yield. R.f. 0.75 (DCM), m.p. 154–156 °C, LCMS: tr = 1.3 min (95% MeOH in water), m/z M-H 409.45, HPLC: tr = 4.7 min (90% acetonitrile in H2O), 98%, 1H NMR (CDCl3, 400 MHz,): δ 1.95 (3H, s, CH3), 4.18 (2H, s, NHCH2), 4.23 (1H, s, NH), 5.93 (2H, s, CH2O), 6.74 (1H, s, ArH), 6.77–6.81 (1H m, ArH), 6.86–6.89 (3H, m, ArH), 7.10 (1H, td, J = 4.0, 8.8 Hz, ArH), 7.24–7.27 (2H, m, ArH), 7.44 (1H, s, ArH), 8.23 (1H, s, ArH). 13C NMR (CDCl3, 101 MHz): δ 24.2 (CH3), 47.2 (CH2NH), 101.4 (CH2O), 105.3, 109.1, 113.4, 118.5, 119.2, 119.4 (ArCH), 121.7 (ArC), 125.4 (ArCH), 128.0 (ArC), 129.8 (ArCH), 131.1, 139.6, 143.7, 144.7, 147.4, 155.9 (ArC), 168.3 (CO). HRMS: Calcd. for C22H19ClN2O4 (M + H)+ 409.0961, found (M + H)+ 409.0957.
(2-Amino-4,5-dimethoxy-phenyl)-methanol
Using the general procedure for the reduction of the substituted 2-nitrobenzylalcohol, the desired compound was obtained as a brown oil, 764 mg, 93% yield. R.f. 0.17 (EtOAc), 1H NMR (CDCl3, 270 MHz,): δ 3.30 (2H, br.s, NH2), 3.78 (3H, s, OCH3), 3.81 (3H, s, OCH3), 4.58 (2H, s, CH2), 6.29 (1H, s, ArH), 6.63 (1H, s, ArH).
N-(2-Hydroxymethyl-4,5-dimethoxy-phenyl)-acetamide
Using the general procedure for the acylation of substituted 2-aminobenzylalcohols, the title compound was obtained as a yellow, waxy solid, 277 mg, 59% yield. R.f. 0.48 (EtOAc), 1H NMR (CDCl3, 270 MHz,): δ 2.14 (3H, s, CH3), 2.85, (1H, br.s, OH), 3.82 (3H, s, OCH3), 3.84 (3H, s, OCH3), 4.56 (2H, s, CH2), 6.69 (1H, s, ArH), 7.46 (1H, s, ArH).
N-(2-[2-(4-Chloro-phenoxy)-phenylamino]-methyl-4,5-dimethoxy-phenyl)-acetamide (5)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the resulting reaction mixture was stirred at r.t. for 2 h, then it was subjected to microwave heating for 5 min at 140 °C. NaHCO3 was added, and the mixture was repeatedly extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography (100% EtOAc) to afford the title compound as a cream solid, 20 mg, 11% yield. R.f. 0.44 (EtOAc), m.p. 117–118 °C, LCMS: tr = 0.97 min (95% MeOH in water), m/z M-H 425.16, HPLC: tr = 1.99 min (90% acetonitrile in H2O), 98%, 1H NMR (CDCl3, 270 MHz,): δ 1.94 (3H, s, CH3), 3.81 (3H, s, OCH3), 3.85 (3H, s, OCH3), 4.22 (3H, br.s, CH2 and NH), 6.74–6.90 (6H, m, ArH), 7.06–7.12 (1H, m, ArH), 7.21–7.26 (2H, m, ArH), 7.57 (1H, s, ArH), 8.26 (1H, br.s, NHCO). 13C NMR (CDCl3, 68 MHz): δ 24.4 (CH3), 47.2 (CH2), 56.11, 56.27 (OCH3), 107.4, 112.5, 113.6, 118.5, 119.3, 119.6 (ArCH), 120.2 (ArC), 125.6 (ArCH), 128.1 (ArC), 129.9, (ArCH), 130.6, 139.9, 143.7, 148.7, 156.0 (ArC), 168.5 (CO). HRMS: Calcd. for C23H23ClN2O4 (M + Na)+ 449.1230, found (M + Na)+ 449.1239.
N-(2-([2-(2,4-Dichloro-phenoxy)-phenylamino]-methyl)-4,5-dimethoxy-phenyl)-acetamide (6)
Here, 2-(2, 4-Dichloro-phenoxy)-phenylamine hydrochloride (135 mg, 0.47 mmol) was dissolved in DCM (10 mL), and K2CO3 (128 mg, 0.94 mmol) was added; the reaction was then stirred at r.t. for 30min. H2O was added to the reaction and the mixture was extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The resulting free amine was dissolved in DCE (2 mL) and to this N-(2-formyl-4,5-dimethoxy-phenyl)-acetamide (69 mg, 0.31 mmol) acetic acid (0.12 mL) and sodium tri-acetoxy borohydride (164 mg, 0.8 mmol) were added. The resulting reaction mixture was stirred at r.t. for 2 h. NaHCO3 was then added, and repeatedly extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography (0–50% EtOAc in hexane) to afford the title compound as an off-white solid, 55 mg, 38% yield. R.f. 0.58 (EtOAc), m.p. 128–131 °C, LCMS: tr = 1.1 min (95% MeOH in water), m/z M-H 459.24, HPLC: tr = 2.2 min (90% acetonitrile in H2O), 96%, 1H NMR (CDCl3, 270 MHz,): δ 1.98 (3H, s, CH3), 3.83 (3H, s, OCH3), 3.87 (3H, s, OCH3), 4.26 (2H, s, CH2), 4.34 (1H, br.s, NH), 6.75–6.77 (3H, m, ArH), 6.83–6.91 (2H, m, ArH), 7.02–7.11 (1H, m, ArH), 7.15 (1H, dd, J = 1.7, 8.7 Hz, ArH), 7.61 (1H, s, ArH), 8.31 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): δ 24.5 (CH3), 47.2 (CH2), 56.1, 56.3 (OCH3), 104.0 (ArC), 107.4, 112.5, 113.6, 118.0, 119.1, 120.0, 125.4 (ArCH), 125.5, 125.8 (ArC), 128.1 (ArCH), 129.0 (ArC), 130.6 (ArCH), 139.1, 143.7, 146.0, 148.7, 151.5 (ArC), 168.5 (CO). HRMS: Calcd. for C23H22Cl2N2O4 (M + H)+ 461.1029, found (M + H)+ 461.1028.
(1-Nitro-naphthalen-2-yl)-methanol
Using the general procedure for the reduction of substituted 2-nitrobenzaldehyde, the desired product was obtained as a dark yellow solid, 1 g, >99% yield. R.f. 0.59 (EtOAc), m.p 78–80 °C, 1H NMR (CDCl3, 270 MHz,): δ 2.32 (1H, br.s, OH), 4.82 (2H, s, CH2), 7.48–7.65 (3H, m, ArH), 7.81–7.90 (2H, m, ArH), 7.99 (1H, d, J = 8.4, ArH).
(1-Amino-naphthalen-2-yl)-methanol
Using the general procedure for the reduction of the substituted 2-nitrobenzylalcohol, the desired product was obtained, 730 mg, 86% yield. R.f. 0.62 (EtOAc), LCMS: tr = 1.3 min (80% MeOH in water), m/z M-H 171.88, HPLC: tr = 2.19 min (70% acetonitrile in water), 87%, 1H NMR (CDCl3, 270 MHz,): δ 4.85 (2H, s, CH2), 7.21–7.25 (2H, m, ArCH), 7.41–7.47 (2H, m, ArH), 7.74–7.85 (2H, m, ArH).
N-(2-Hydroxymethyl-naphthalen-1-yl)-acetamide
Using the general procedure for the acylation of substituted 2-aminobenzylalcohols, the desired product was obtained as a yellow solid, 377 mg, 82% yield. R.f. 0.58 (EtOAc), m.p. 105–108 °C, HPLC tr = 1.45 min (90% acetonitrile in H2O) >99%, LCMS tr = 0.92 min (95% MeOH in H2O) M + Na 237.90, 1H NMR (CDCl3, 270 MHz,): δ 2.32 (3H, s, CH3), 3.29 (1H, br.s, OH), 4.63 (2H, s, CH2), 7.40–7.44 (3H, m, ArH), 7.77–7.80 (3H, m, ArH).
N-(2-[2-(4-Chloro-phenoxy)-phenylamino]-methyl-naphthalen-1-yl)-acetamide (7)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated, 28 mg, 24% yield. R.f. 0.38 (EtOAc), m.p. 153–155 °C, LCMS: tr = 1.14 min (95% MeOH in water), m/z M-H 415.34, HPLC: tr = 2.26 min (90% acetonitrile in water), 96%, 1H NMR (CDCl3, 270 MHz,): δ 2.28 (3H, s, CH3), 4.42 (2H, s, CH2), 4.53 (1H, br.s, NH), 6.63–6.73 (2H, m, ArH), 6.83–6.91 (3H, m, ArH), 6.97–7.03 (1H, m, ArH), 7.21–7.24 (2H, m, ArH), 7.42–7.51 (4H, m, ArH), 7.75 (1H, d, J = 8.4 Hz, ArH), 7.80–7.85 (2H, m, ArH and NHCO). 13C NMR (CDCl3, 68 MHz): 23.5 (CH3), 45.4 (CH2), 112.4, 117.7, 118.5, 119.6, 122.6, 125.5, 126.0, 126.1 (ArCH), 127.8 (ArC), 128.2, 128.4, 129.8 (ArCH), 130.5, 130.6, 133.4, 133.7, 140.2, 142.9, 156.5 (ArC), 169.4 (CO). HRMS: Calcd. for C25H21ClN2O2 (M + H)+ 439.1184, found (M + H)+ 439.1190.
(2-Amino-5-methyl-phenyl)-methanol
Using the general procedure for the reduction of the substituted 2-nitrobenzylalcohol, the desired compound was obtained as a brown solid, 162 mg, 79% yield. R.f. 0.45 (EtOAc), m.p. 118–121 °C, LCMS: tr = 0.93 min (95% MeOH in water), m/z M + H 137.80, HPLC: tr = 1.53 min (90% acetonitrile in H2O), 96%, 1H NMR (CDCl3, 270 MHz,): δ 2.22 (3H, s, CH3), 4.02 (2H, br.s, NH), 4.64 (2H, s, CH2), 6.62 (1H, d, J = 9.3 Hz, ArH), 6.89–6.95 (2H, m, ArH), 7.25 (1H, s, OH).
N-(2-Hydroxymethyl-4-methyl-phenyl)-acetamide
Using the general procedure for the acylation of substituted 2-aminobenzylalcohols, the title compound was obtained as a cream solid, 180 mg, 79% yield. R.f. 0.35 (EtOAc), m.p. 134–136 °C (from hexane), LCMS tr = 1.4 min (80% MeOH in H2O) M + Na 210.99, 1H NMR (CDCl3, 270 MHz,): δ 2.17 (3H, s, CH3), 2.29 (3H, s, CH3), 4.63 (2H, d, J = 4.9 Hz, CH2), 7.02 (1H, s, ArH), 7.12 (1H, d, J = 8.4 Hz, ArH), 7.80 (1H, d, J = 8.2 Hz, ArH), 8.28 (1H, s, NH).
N-(2-[2-(4-Chloro-phenoxy)-phenylamino]-methyl-4-methyl-phenyl)-acetamide (8)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a light cream solid, 37 mg, 47% yield. R.f. 0.35 (EtOAc), m.p. 138–140 °C (from hexane), LCMS: tr = 1.21 min (95% MeOH in water), m/z M + H 381.20, HPLC: tr = 2.38 min (90% acetonitrile in H2O), 96%. 1H NMR (CDCl3, 270 MHz,): δ 1.97 (3H, s, CH3), 2.29 (3H, s, CH3), 4.25 (3H, s, CH2 and NH), 6.76–6.93 (5H, m, ArCH), 7.07–7.13 (3H, m, ArH), 7.22–7.27 (2H, m, ArH), 7.83 (1H, d, J = 8.2 Hz, ArH), 8.35 (1H, br.s, NH). 13C NMR (CDCl3, 68 MHz): δ 20.9, 24.5 (CH3), 47.5 (CH2), 113.6, 118.6, 119.3, 119.6, 123.1, 125.5 (ArCH), 128.0, 128.1 (ArC), 129.3, 129.9, 130.2 (ArCH), 134.4, 134.7, 139.9, 143.8, 156.0 (ArC), 168.5 (CO). HRMS: Calcd. for C22H21ClN2O2 (M + H)+ 381.1364, found (M + H)+ 381.1365.
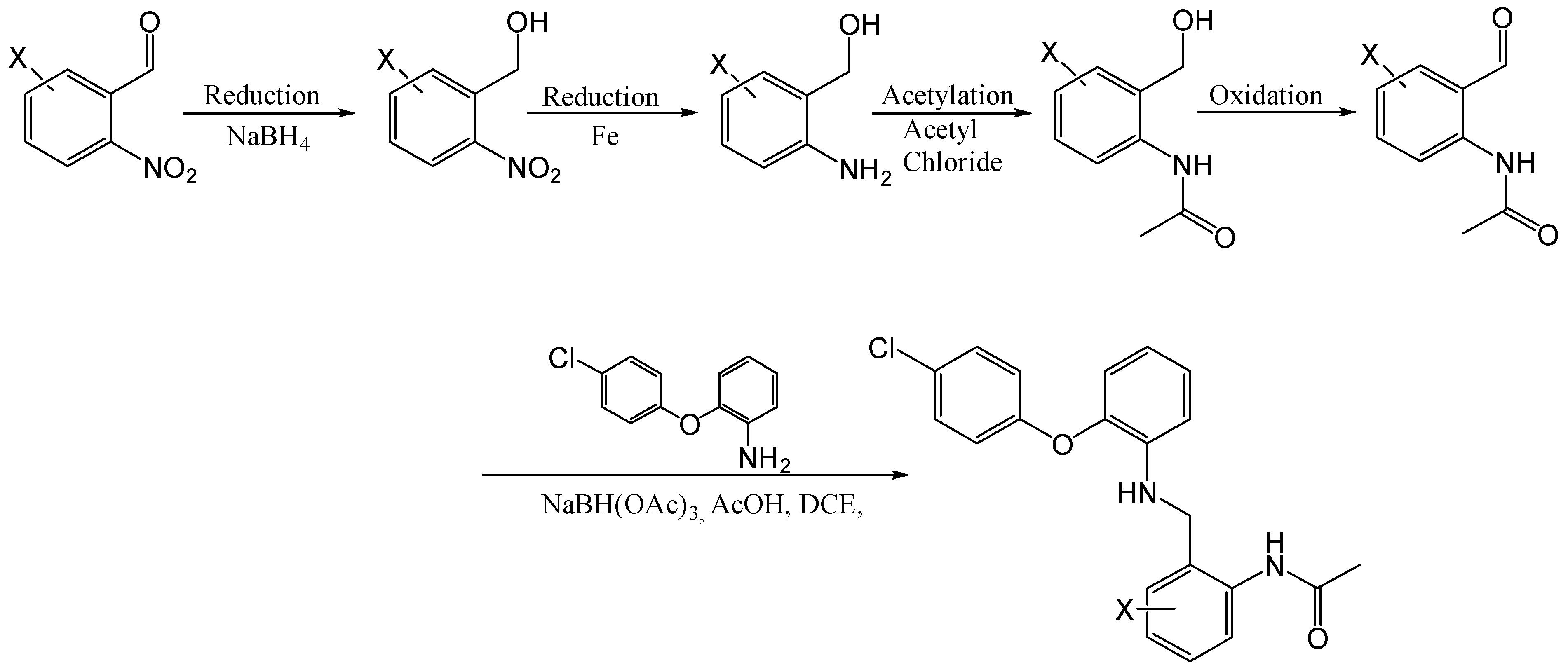
N-(4-Chloro-2-hydroxymethyl-phenyl)-acetamide
Using the general procedure for the acylation of substituted 2-aminobenzylalcohols, the title compound was obtained as a cream wax, 1.12 g, 89% yield. R.f. 0.35 (EtOAc), 1H NMR (CDCl3, 270 MHz,): δ 1.46 (2H, s, NH and OH), 2.19 (3H, s, CH3), 4.60 (2H, s, CH2), 7.14–7.26 (2H, m, ArH), 7.94–7.98 (1H, m, ArH).
N-(4-Chloro-2-[2-(4-chloro-phenoxy)-phenylamino]-methyl-phenyl)-acetamide (9)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a light brown wax, 68mg, 26% yield. R.f. 0.43 (1:1, EtOAc: Hexane), LCMS: tr = 1.21 min (95% MeOH in water), m/z M-H 399.15, 401.1, HPLC: tr = 2.62 min (90% acetonitrile in H2O), 97%, 1H NMR (CDCl3, 270 MHz,): δ 1.95 (3H, s, CH3), 4.22 (3H, s, NH and CH2), 6.79–6.91 (5H, m, ArH), 7.07–7.13 (1H, m, ArH), 7.24–7.28 (4H, m, ArH), 7.98 (1H, d, J = 8.4 Hz, ArH), 8.56 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): δ 24.5 (CH3), 47.5 (CH2), 113.8, 118.7, 119.5, 119.9, 124.1, 125.5, 128.7, 129.3 (ArCH), 129.5 (ArC), 129.9 (ArCH), 136.0, 139.4, 144.1, 155.9, 168.5, 200.5 (ArC), 205.1 (CO). HRMS: Calcd. for C21H18Cl2N2O2 (M + H)+ 401.0818, found (M + H)+ 401.0803.
N-(4-Chloro-2-[2-(2,4-dichloro-phenoxy)-phenylamino]-methyl-phenyl)-acetamide (10)
Here, 2-(2,4-Dichloro-phenoxy)-phenylamine hydrochloride (350 mg, 1.22 mmol) was dissolved in DCM (10 mL), and K2CO3 (335 mg, 2.44 mmol) was added; the reaction was then stirred at r.t. for 30 min. H2O was added and the mixture was extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The resulting amine was dissolved in DCE (2 mL) and N-(4-chloro-2-formyl-phenyl)-acetamide (160 mg, 0.81 mmol), acetic acid (0.15 mL) and sodium tri-acetoxy borohydride (0.43 g, 2.02 mmol) were added. The resulting reaction mixture was stirred at r.t. for 2 h. NaHCO3 was added and the mixture was repeatedly extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography (0–50% EtOAc in hexane) to afford the title compound as a cream solid, 210 mg, 60% yield. R.f. 0.38 (EtOAc), m.p. 135–137 °C, LCMS: tr = 1.3 min (95% MeOH in water), m/z M-H 433.1, 435.1, HPLC: tr = 2.8 min (90% acetonitrile in H2O), 99%, 1H NMR (CDCl3, 270 MHz,): δ 1.98 (3H, s, CH3), 4.28–4.29 (2H, m, CH2), 4.40 (1H, br.s, NH), 6.70–6.88 (4H, m, ArH), 7.04–7.10 (1H, m, ArH), 7.17 (1H, dd, J = 2.5 Hz, ArH), 7.25–7.27 (2H, m, ArH), 7.45 (1H, d, J = 2.5 Hz, ArH), 7.96–7.99 (1H, m, ArCH), 8.64 (1H, br.s. NH). 13C NMR (CDCl3, 101 MHz): δ 24.6 (CH3), 47.4 (CH2), 113.8, 117.8, 119.7, 120.3, 124.1, 125.3 (ArCH), 125.9 (ArC), 128.2, 128.7, 129.3 (ArCH), 129.4, 129.6 (ArC), 130.7 (ArCH), 136.0, 138.6, 144.4, 151.1 (ArC), 168.5 (CO). HRMS: Calcd. for C21H17Cl3N2O2 (M + H)+ 435.0428, found (M + H)+ 437.0387.
N-(4-Chloro-2-[2-(4-trifluoromethoxy-phenoxy)-phenylamino]-methyl-phenyl)-acetamide (11)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a brown solid, 86 mg, 30% yield. R.f. 0.35 (1:1, EtOAc: Hexane), m.p. 121–123 °C, LCMS: tr = 1.07 min (95% MeOH in water), m/z M-H 449.29, HPLC: tr = 3.28 min (90% acetonitrile in H2O), >99%, 1H NMR (CDCl3, 270 MHz,): δ 1.94 (3H, s, CH3), 4.23 (3H, s, CH2 and NH), 6.80–6.96 (5H, m, ArH), 7.08–7.18 (3H, m, ArH), 7.25–7.30 (3H, m, ArH), 7.99 (1H, dd, J = 8.4 Hz, ArH), 8.54 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): 24.4 (CH3), 47.4 (CH2), 113.8, 118.1, 119.6, 119.8, 112.8, 123.9, 125.5, 128.6, 129.2 (ArCH), 135.9, 139.3 (ArC), 155.6 (OCF3), 207.9 (CO). 19F NMR (CDCl3, 376 MHz): δ −58.3 (OCF3). Anal. Calcd. for C22H18ClF3N2O3: C 58.61, H 4.02, N 6.21%. Found: C 58.1, H 4.12, N 5.96%. HRMS: Calcd. for C22H18ClF3N2O3 (M + H)+ 451.1031, found (M + H)+ 451.1024.
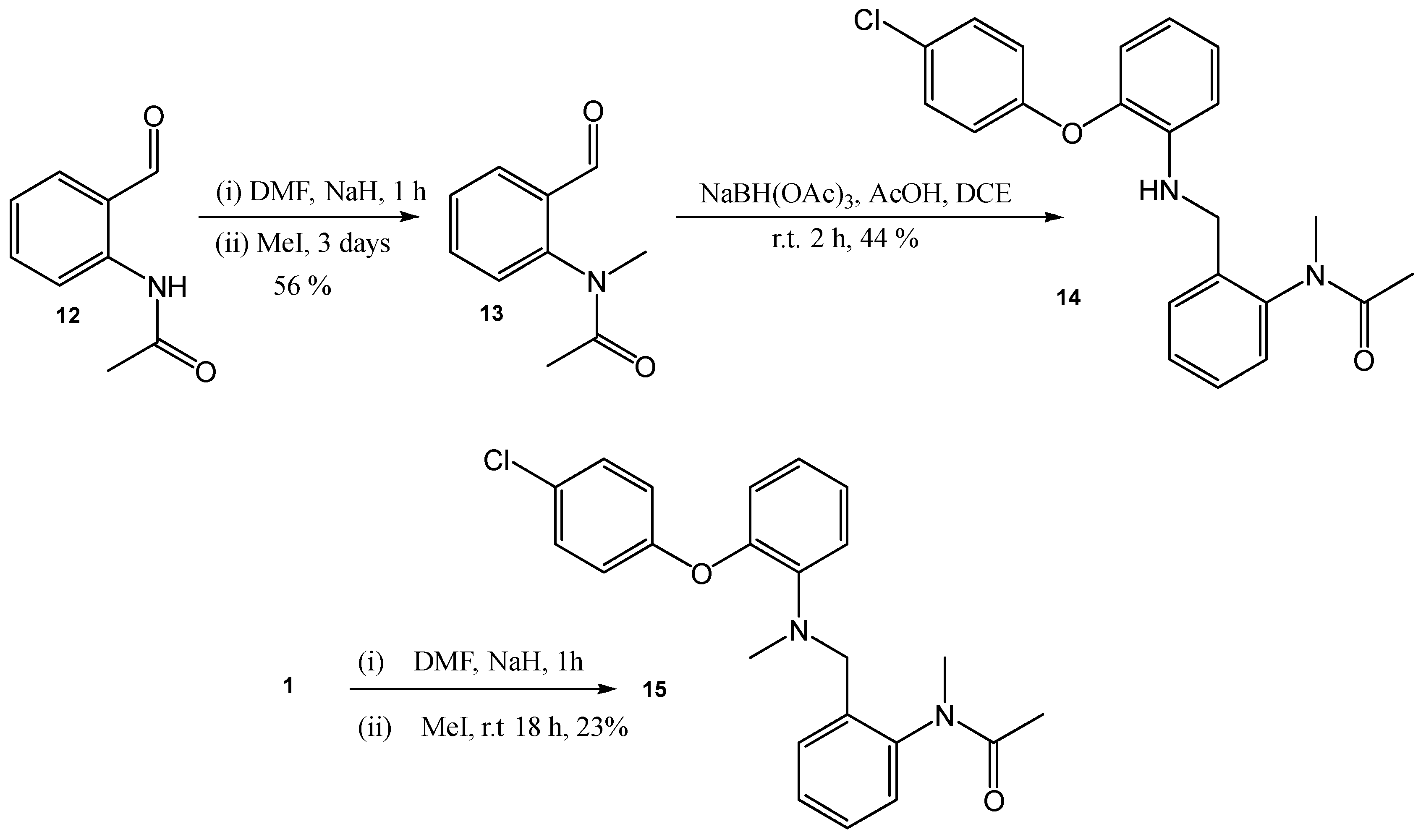
N-(2-([2-(4-Chloro-phenoxy)-phenylamino]-methyl)-phenyl)-N-methyl-acetamide (14)
To a solution of 2-(4-chloro-phenoxy)-phenylamine (174 mg, 0.78 mmol) and N-(2-formyl-phenyl)-N-methyl-acetamide 13 (70 mg, 0.39 mmol) in DCE (2mL), NaHB(OAc)3 (210 mg, 0.98 mmol) and AcOH (0.07 mL) were added. The resulting solution was stirred at r.t. for 2 h. NaHCO3 was then added, and the mixture was extracted with DCM and dried (MgSO4). The crude product was purified by flash chromatography (0–100% DCM in hexane) to yield the desired product as a brown oil, 65 mg, 44% yield. R.f. 0.51 (DCM), LCMS: tr = 1.19 min (95% MeOH in water), m/z M-H 378.93, HPLC: tr = 2.64 min (90% acetonitrile in water), 98%, 1H NMR (CDCl3, 270 MHz,): δ 1.78 (3H, s, CH3CO), 3.24 (3H, s, CH3N), 4.25–4.29 (2H, m, CH2), 6.59 (1H, dd, J = 1.4, 8.0 Hz, ArH), 6.67 (1H, td, J = 1.4, 7.7 Hz, ArH), 6.83 (1H, dd, J = 1.4, 7.7 Hz, ArH), 6.88–6.92 (2H, m, ArH), 7.00 (1H, td, J = 1.6, 7.7 Hz, ArH), 7.11–7.17 (1H, m, ArH), 7.23–7.26 (2H, m, ArH), 7.31–7.34 (2H, m, ArH), 7.41–7.44 (1H, m, ArH). 13C NMR (CDCl3, 68 MHz): 26.5, 36.6 (CH3), 43.7 (CH2), 117.7, 118.7, 119.5, 125.4 (ArCH), 127.9 (ArC), 128.5, 128.9, 129.1, 129.7 (ArCH), 136.3, 139.9, 142.5, 142.8, 156.1 (ArC), 170.8 (CO). HRMS: Calcd. for C22H21ClN2O2 (M + H)+ 381.1364, found (M + H)+ 381.1378.
N-[2-(([2-(4-Chloro-phenoxy)-phenyl]-methyl-amino)-methyl)-phenyl]-N-methyl-acetamide (15)
N-(2-([2-(4-chloro-phenoxy)-phenylamino]-methyl)-phenyl)-acetamide (100 mg, 0.27 mmol) in DMF (10 mL) was cooled to 0 °C, NaH (35 mg, 0.81 mmol) was added and the resulting solution was stirred for 1 h. MeI (0.05 mL, 0.81 mmol) was added, and the solution stirred for a further 18 h. The reaction mixture was then poured onto water, extracted with EtOAc and dried (MgSO4). NMR analysis showed the crude product to be a mixture of product and the related mono-methylated compound. Preparative HPLC was used for purification to yield the desired mono-methylated product as a white wax, 25 mg, 23% yield. Please note N-(2-([2-(4-chloro-phenoxy)-phenylamino]-methyl)-phenyl)-N-methyl-acetamide was also isolated, 46 mg, 44% yield. R.f. 0.45 (EtOAc), LCMS: tr = 5.6 min (80% MeOH in water), m/z M + H 395.18, HPLC: tr = 3.75 min (90% acetonitrile in water), 98%, 1H NMR (CDCl3, 270 MHz,): δ 1.70 (3H, s, CH3), 2.66 (3H, s, NCH3), 3.09 (3H, s, NCH3), 4.10 (2H, s, CH2), 6.67–6.73 (2H, m, ArH), 6.96 (2H, d, J = 3.9 Hz, ArH), 7.04–7.27 (8H, m, ArH). 13C NMR (CDCl3, 68 MHz): δ 22.0, 36.3, 39.7 (CH3), 54.9 (CH2), 117.8, 119.7, 122.2, 122.5, 125.6 (ArCH), 127.1 (ArC), 142.4, 145.0, 147.4 (ArC), 170.8 (CO). HRMS: Calcd. for C23H23ClN2O2 (M + H)+ 395.1521, found (M + H)+ 395.1533.
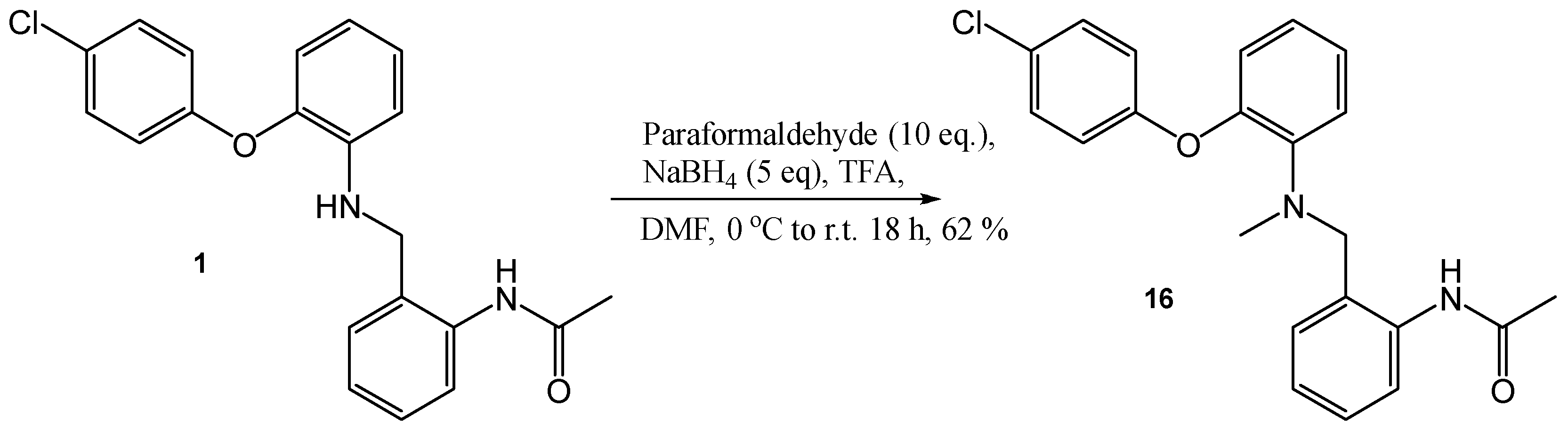
N-[2-(([2-(4-Chloro-phenoxy)-phenyl]-methyl-amino)-methyl)-phenyl]-acetamide (16)
To a solution of N-(2-[2-(4-chloro-phenoxy)-phenylamino]-methyl-phenyl)-acetamide, (100 mg, 0.27 mmol), paraformaldehyde (81 mg, 2.7 mmol) and NaBH4 (55 mg, 1.35 mmol) in THF (5 mL) were added TFA (1.3 mL). The resulting solution was stirred at r.t. for 18 h. This was then poured into NaOH solution (25%) with ice chips, extracted with DCM and dried (MgSO4). The crude product was then purified by flash chromatography (0–100% EtOAc in hexane) to produce the desired compound as a colourless oil, 64 mg, 62% yield. R.f. 0.66 (EtOAc), LCMS: tr = 4.02 min (80% MeOH in water), m/z M + H 379.12, HPLC: tr = 2.88 min (90% MeOH in water), 99%, 1H NMR (CDCl3, 270 MHz,): δ 1.99 (3H, s, CH3), 2.65 (3H, s, CH3N), 4.19 (2H, s, CH2), 6.81 (1H, dd, J = 1.6, 8.4 Hz, ArH), 6.93–6.95 (2H, m, ArH), 6.99–7.04 (2H, m, ArH), 7.10 (1H, td, J = 1.6, 8.0 Hz, ArH), 7.14–7.15 (1H, m, ArH), 7.23–7.32 (4H, m, ArH), 8.27 (1H, d, J = 8.4 Hz, ArH), 10.12 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): 24.8, 40.7 (CH3), 59.5 (CH2), 118.7, 120.3, 120.9, 121.3, 123.2, 124.1, 124.6, (ArCH), 125.1 (ArC), 128.6 (ArC), 130.1 (ArCH), 138.6, 142.4, 150.8, 155.1 (ArC), 168.6 (CO). HRMS: Calcd. for C22H21ClN2O2 (M + H)+ 381.1364, found (M + H)+ 381.1363.
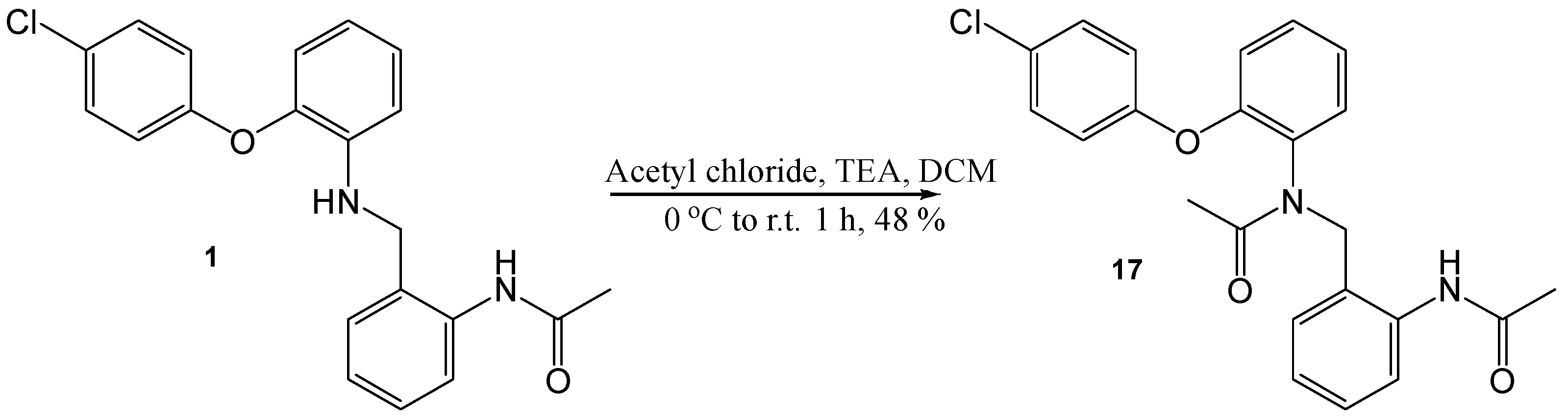
N-(2-Acetylamino-benzyl)-N-[2-(4-chloro-phenoxy)-phenyl]-acetamide (17)
A solution of N-(2-[2-(4-chloro-phenoxy)-phenylamino]-methyl-phenyl)-acetamide (100 mg, 0.27 mmol) in DCM (5 mL) was cooled to 0 °C, and TEA (0.2 mL) and acetyl chloride (0.34 mL, 0.81 mmol) were added; the resulting solution was stirred at r.t. for 1 h. Saturated NaHCO3 was added, extracted with DCM and dried (MgSO4). The crude product was purified by flash chromatography (0–100% EtOAc in hexane) and preparative HPLC to yield the desired product as an off white waxy solid, 53 mg, 48% yield. R.f. 0.54 (EtOAc), LCMS: tr = 2.17 min (95% MeOH in water), m/z M-H 407.15, HPLC: tr = 2.40 min (90% acetonitrile in water), 95%, 1H NMR (CDCl3, 270 MHz,): δ 1.93 (3H, s, CH3), 2.24 (3H, s, CH3), 4.80 (2H, s, CH2), 7.02–7.12 (2H, m, ArH), 7.21–7.30 (4H, m, ArH), 8.20 (1H, dd, J = 7.7 Hz, ArH), 9.89 (1H, s, NHCO). 13C NMR (CDCl3, 68 MHz): 22.1, 24.6 (CH3), 49.8 (CH2), 118.1, 120.7, 122.1, 123.0, 124.0 (ArCH), 125.0 (ArC), 129.2 (ArCH), 129.8 (ArC), 130.1, 130.2, 131.5 (ArCH), 137.7, 153.2, 153.8 (ArC), 169.5, 172.7 (CO). HRMS: Calcd. for C23H21ClN2O3 (M + Na)+ 431.1313, found (M + Na)+ 431.1105.
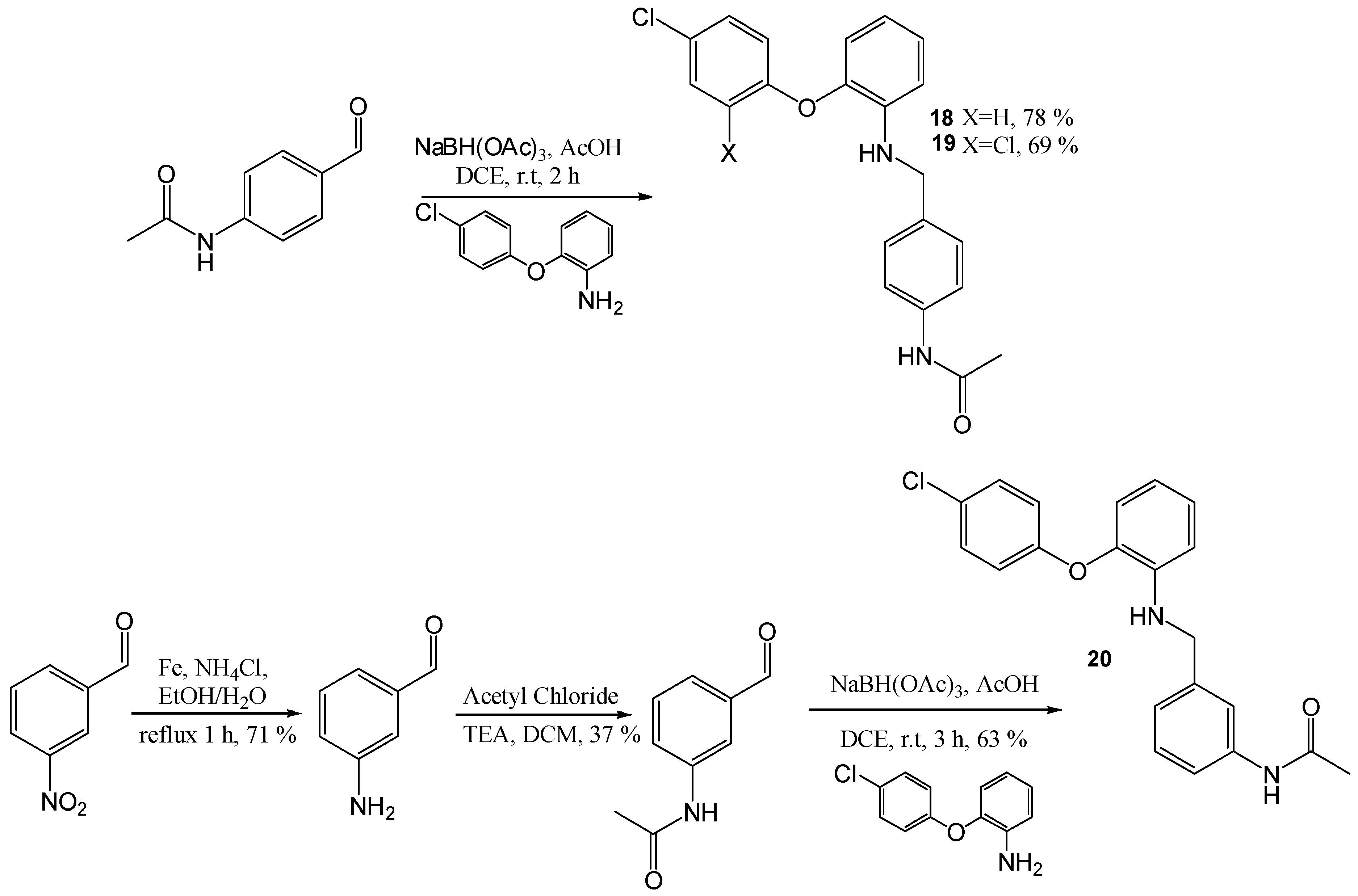
N-(4-([2-(4-Chloro-phenoxy)-phenylamino]-methyl)-phenyl)-acetamide (18)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline 4-acetamido-benzaldehyde, the desired compound was isolated as a light pink solid, 195 mg, 78% yield. R.f. 0.55 (5% MeOH in DCM), m.p. 137–139 °C, LCMS: tr = 1.39 min (95% MeOH in H2O), m/z M-H 365.48, HPLC: tr = 2.0 min (90% acetonitrile in H2O), 98%, 1H NMR (CDCl3, 270 MHz): δ 2.15 (3H, s, CH3), 4.30 (2H, d, J = 5.7 Hz, CH2), 6.60–6.69 (2H, m, ArH), 6.82 (1H, dd, J = 1.5, 7.7 Hz, ArH), 6.86–6.91 (2H, m, ArH), 6.96–7.02 (1H, m, ArH), 7.12 (1H, s, NH), 7.21–7.26 (4H, m, ArH), 7.41–7.44 (2H, m, ArH). 13C NMR (CDCl3, 101 MHz): δ 24.6 (CH3), 47.2 (CH2), 111.9, 117.1, 118.5, 119.3, 120.1, 125.3 (ArCH), 127.6 (ArC), 127.9, 129.6 (ArCH), 135.1, 136.8, 140.1, 142.6, 156.2 (ArC), 168.2 (CO). HRMS: Calcd. for C21H19ClN2O2 (M + Na)+ 389.1025, found (M + Na)+ 389.1028. Anal. Calcd. for C21H19ClN2O2: C 68.76, H 5.22 N 7.64%. Found: C 68.5, H 5.26, N 7.61%.
N-(4-([2-(2,4-Dichloro-phenoxy)-phenylamino]-methyl)-phenyl)-acetamide (19)
To a solution of 2-(2,4-dichloro-phenoxy)-phenylamine hydrochloride (0.15 g, 0.52 mmol) in DCM (10 mL), K2CO3 (0.22 g, 1.04 mmol) was added,;the resulting solution was stirred at r.t for 30 min. Water was added and the mixture was extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The resulting amine was dissolved in DCE (3 mL) and 4-acetamidobenzaldehyde (0.126 g, 0.0.75 mmol), acetic acid (0.11 mL) and sodium tri-acetoxy borohydride (0.27 g, 1.3 mmol) were added. The resulting reaction mixture was stirred at r.t. for 2 h. NaHCO3 was added, and the mixture was repeatedly extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography (0–100% EtOAc in hexane) to afford the title compound as a white wax, 142 mg, 69% yield. R.f. 5.8 (EtOAc), LCMS: tr = 1.2 min (95% MeOH in H2O), m/z M-H 399.03, 401.04, HPLC: tr = 2.42 min (90% acetonitrile in H2O), 98%, 1H NMR (CDCl3, 270 MHz): δ 2.15 (3H, s, CH3), 4.31 (2H, s, CH2), 4.59 (1H, br.s, NH), 6.59–6.68 (2H, m, ArH), 6.75 (1H, dd, J = 1.5, 7.9 Hz, ArH), 6.80 (1H, d, J = 8.7 Hz, ArH), 6.96–7.02 (1H, m, ArH), 7.12 (1H, dd, J = 2.5, 8.9 Hz, ArH), 7.22–7.31 (1H, m, ArH), 7.41–7.45 (2H, m, ArH). 13C NMR (CDCl3, 68 MHz): δ 24.7 (CH3), 47.3 (CH2), 112.2, 117.1, 118.5, 119.4, 120.2 (ArCH), 125.3 (ArC), 125.5, 127.9, 128.0 (ArCH), 128.2 (ArC), 130.4 (ArCH), 135.1, 136.9, 139.7, 142.7, 151.8 (ArC), 168.4 (CO). HRMS: Calcd. for C21H18Cl2N2O2 (M + H)+ 399.0673, found (M + H)+ 399.0674. Anal. Calcd. for C21H18Cl2N2O2 C 62.85, H 4.52 N 6.98%. Found: C 62.7, H 4.52, N 6.92%.
3-Amino-benzaldehyde
Using the general procedure for the reduction of the substituted 2-nitrobenzylalcohol, the desired compound was obtained as a yellow solid, 1.7 g, 71% yield. R.f. 0.25 (DCM). Due to the instability of this compound, the product was used crude in the following reactions.
N-(3-[2-(4-Chloro-phenoxy)-phenylamino]-methyl-phenyl)-acetamide (20)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline 3-acetamide benzaldehyde, the desired compound was isolated as a cream solid, 110 mg, 63% yield. R.f. 0.6 (EtOAc), m.p. 187–190 °C, LCMS: tr = 1.39 min (50% to 95% MeOH in water at 0.5 mL/min to 1.0 mL/min over 5 min), m/z M-H 365.55, HPLC: tr = 1.89 min (90% acetonitrile in H2O), 93%, 1H NMR (CDCl3, 270 MHz,): δ 2.13 (3H, s, CH3), 4.32 (2H, d, J = 5.4 Hz, CH2), 4.55 (1H, d, J = 5.4 Hz, NH), 6.61–6.66 (2H, m, ArH), 6.81–6.84 (1H, m, ArH), 6.68–6.92 (2H, m, ArH), 6.98–7.04 (2H, m, ArH), 7.21–7.27 (4H, m, ArH and NH), 7.38–7.43 (2H, m, ArH). 13C NMR (CDCl3, 68 MHz): δ 24.6 (CH3), 47.5 (CH2), 112.1, 117.2, 118.4, 118.7, 119.4, 123.0, 125.4 (ArCH), 127.7 (ArC), 129.0, 129.7, 129.9 (ArC), 138.2, 140.1, 140.4, 142.7, 156.3 (ArC), 168.5 (CO). HRMS: Calcd. for C21H19ClN2O2 (M + Na)+ 389.1027, found (M + Na)+ 389.1021.
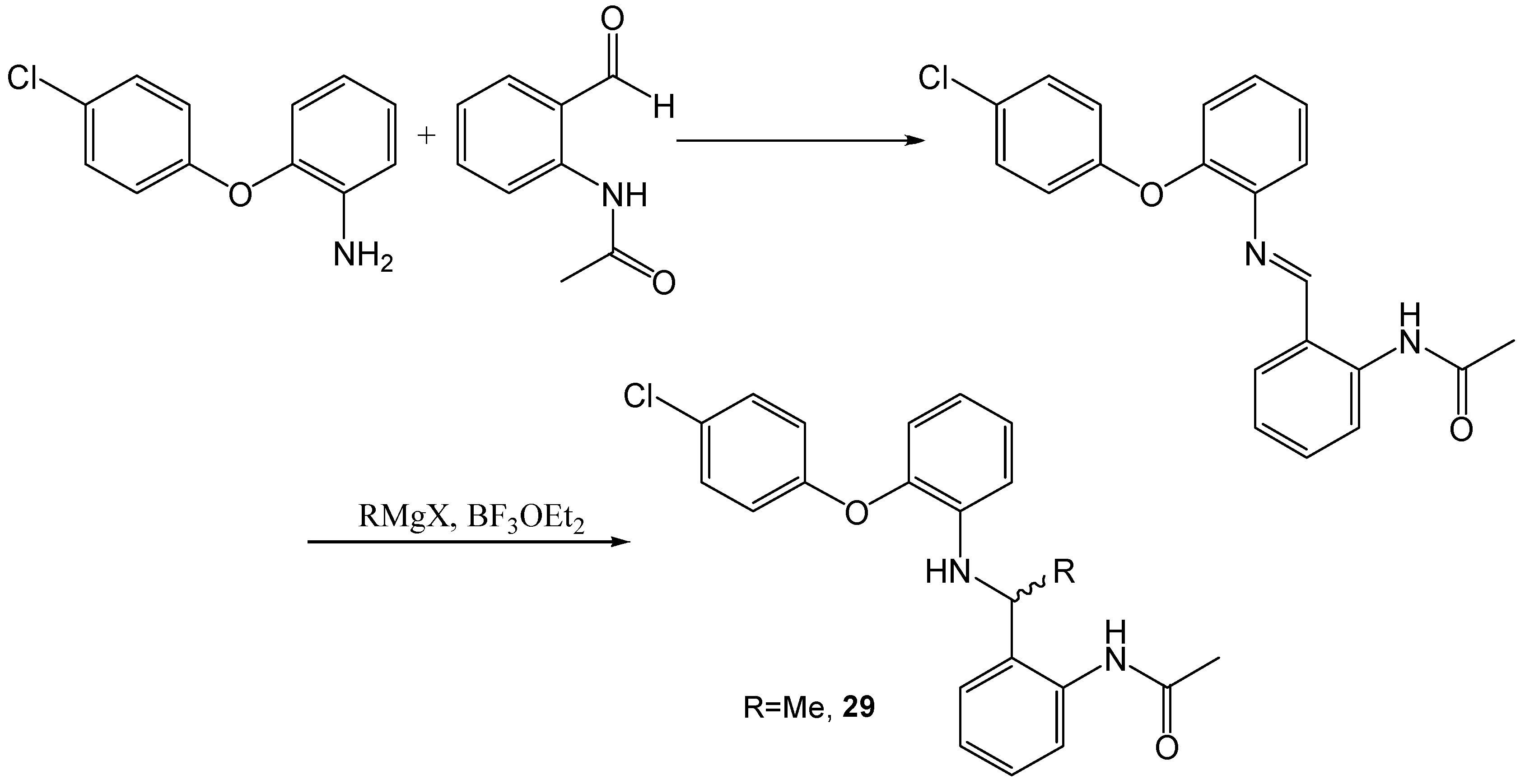
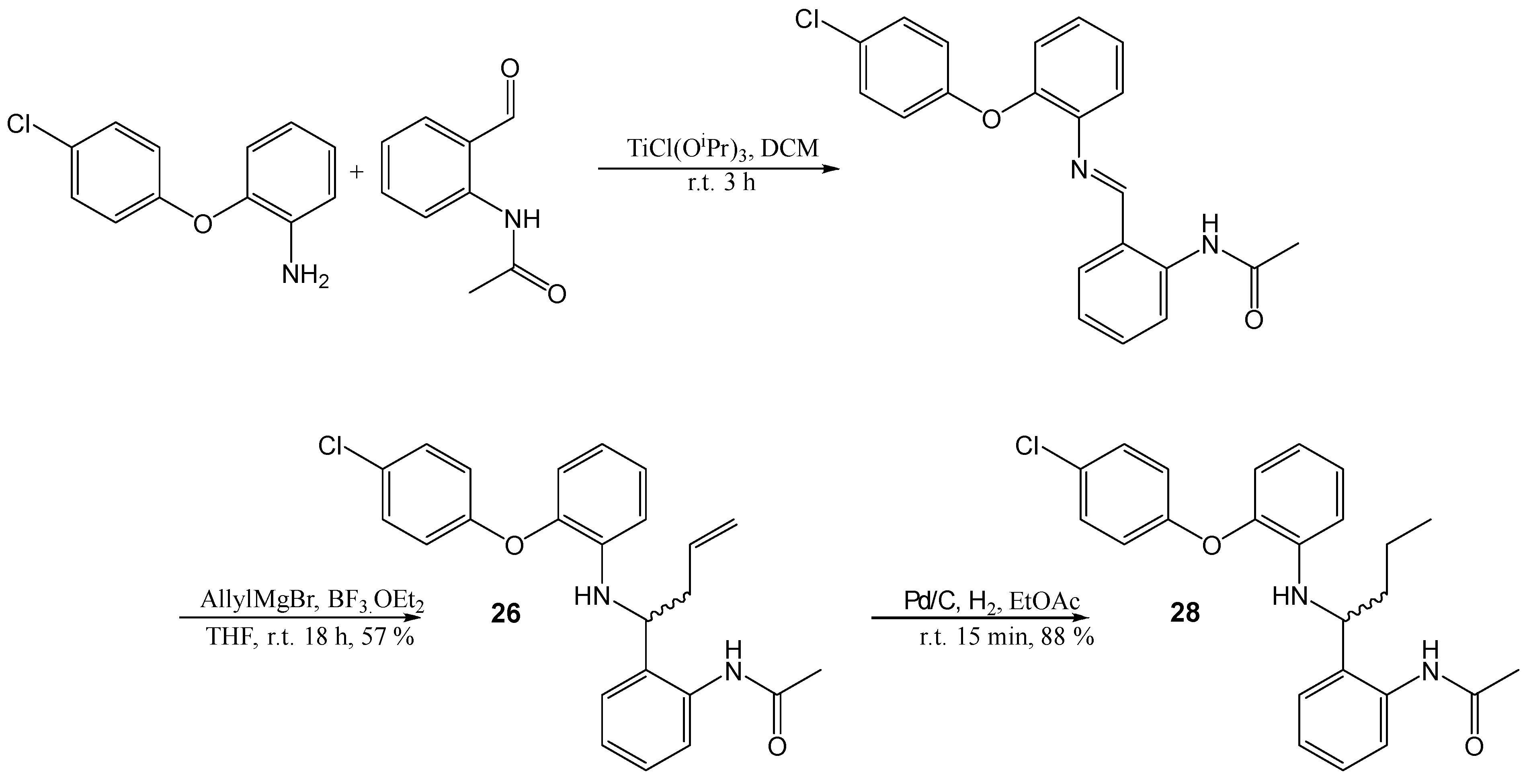
N-(2-([2-(4-Chloro-phenoxy)-phenylimino]-methyl)-phenyl)-acetamide
A solution of 2-(4-chloro-phenoxy)-phenylamine (100 mg, 0.46 mmol) and N-(2-formyl-phenyl)-acetamide 12 (74 mg, 0.46 mmol) in anhydrous DCM (5mL) was stirred at r.t., MgSO4 (550 mg, 4.6 mmol) was added and the resulting mixture stirred for a further 18 h at r.t. The mixture was then filtered and the solid was washed with DCM. The filtrate was then evaporated to dryness to yield the desired product as an oil. The product was identified by NMR, as no CHO peak was visible in the 1H NMR and it had been replaced with an imine peak. The product was used crude in all following experiments. A small sample was purified for analytical purposes. 1H NMR (CDCl3, 270 MHz,): δ 2.03 (3H, s, CH3), 6.87–7.46 (11H, m, ArH), 8.60 (1H, s, N=CH), 8.72 (1H, d, J = 8.5 Hz, ArH). 13C NMR (CDCl3, 101 MHz): 24.9 (CH3), 116.7, 119.2, 119.6, 120.3 (ArCH), 120.6 (ArC), 122.5, 125.0, 127.9 (ArCH), 128.2 (ArC), 129.8, 132.7 (ArCH), 140.4, 141.7, 149.6, 156.1 (ArC), 163.4 (CH), 169.9 (CO).
N-(2-([2-(4-Chloro-phenoxy)-phenylimino]-methyl)-phenyl)-acetamide
A solution of 2-(4-chloro-phenoxy)-phenylamine (100 mg, 0.46 mmol) and N-(2-formyl-phenyl)-acetamide 12 (74 mg, 0.46 mmol) in anhydrous DCM (5 mL) was stirred at r.t., and TiCl(OiPr)3 (0.25 mL, 1 mmol) was added. The resulting mixture was stirred for a further 4 h at room temperature. The mixture was then evaporated to dryness to yield the desired product as an oil. The product was used crude in all subsequent experiments.
[2-(4-Chloro-phenoxy)-phenyl]-2-nitro-benzylamine
To a solution of 2-(4-chloro-phenoxy)-phenylamine (150 mg, 0.68 mmol) and 2-nitrobenzaldehyde (310 mg, 2.04 mmol) in DCE (3.5 mL), acetic acid (0.36 mL) and sodium tri-acetoxy borohydride (0.36 g, 1.7 mmol) were added. The resulting reaction mixture was heated in a microwave at 140 °C for 10 min. NaHCO3 was then added and the mixture was repeatedly extracted with EtOAc. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography (0–100% EtOAc in hexane) to afford the title compound as a yellow wax, 194 mg, 77% yield. R.f. 0.63 (1:1, EtOAc: Hexane), LCMS: tr = 1.66 min (95% MeOH in H2O), m/z M + H 355.48, HPLC: tr = 6.6 min (90% acetonitrile in H2O), 92%, 1H NMR (CDCl3, 270 MHz): δ 4.75 (2H, s, CH2), 4.97 (1H, s, NH), 6.52 (1H, dd, J = 1.2, 7.9 Hz, ArH), 6.66 (1H, td, J = 1.5, 7.7 Hz, ArH), 6.83–6.99 (4H, m, ArH), 7.21–7.27 (2H, m, ArH), 7.37–7.44 (1H, m, ArH), 7.54–7.57 (2H, m, ArH), 8.05 (1H, dd, J = 1.0, 7.7 Hz, ArH).
N-[2-(4-Chloro-phenoxy)-phenyl]-N-(2-nitro-benzyl)-acetamide
Using the general procedure for the acylation of substituted 2-aminobenzylalcohols, the title compound was obtained as a brown oil, 92 mg, 48% yield. R.f. 0.21 (1:1, DCM:hexane), LCMS: tr = 5.12 min (50% to 95% MeOH in water at 0.5 mL/min to 1.0 mL/min over 5 min), m/z M + H 397.48, HPLC: tr = 5.0 min (90% acetonitrile in H2O), 91%, 1H NMR (CDCl3, 270 MHz,): δ 1.98 (3H, s, CH3), 5.13 (1H, d, J = 16.3 Hz, ½CH2), 5.33 (1H, d, J = 16.3 Hz, ½CH2), 6.79–6.87 (3H, m, ArH), 7.00–7.11 (2H, m, ArH), 7.20–7.35 (4H, m, ArH), 7.46 (1H, td, J = 1.5, 7.4 Hz, ArH), 7.72 (1H, dd, J = 1.2, 7.9 Hz, ArH), 7.83 (1H, dd, J = 1.2, 8.2 Hz, ArH).
N-(2-Amino-benzyl)-N-[2-(4-chloro-phenoxy)-phenyl]-acetamide
Using the general procedure for the reduction of the substituted 2-nitrobenzylalcohol, the desired compound was obtained as a pale-yellow solid, 53 mg, 62% yield. R.f. 0.25 (1:1, DCM:EtOAc), 1H NMR (CDCl3, 270 MHz,): δ 1.92 (3H, s, CH3), 4.55 (2H, br.s, NH2), 4.73–4.87 (2H, m, CH2), 6.38–6.52 (3H, m, ArH), 6.58–6.64 (2H, m, ArH), 6.77 (1H, d, J = 8.2 Hz, ArH), 6.96–7.05 (3H, m, ArH), 7.19–7.25 (3H, m, ArH). 13C NMR (CDCl3, 68 MHz): δ 22.2 (CH3), 49.3 (CH2), 115.41, 116.7, 118.3 (ArCH), 119.8 (ArC), 120.7, 123.8, 129.3, 129.6, 129.9, 130.4, 131.9 (ArCH), 132.1, 146.5, 153.4, 154.4 (ArC), 171.7 (CO).
[2-(4-Chloro-phenoxy)-phenyl]-(2-amino-benzyl)-amine
Using the general procedure for the reduction of the substituted 2-nitrobenzylalcohol, the desired compound was obtained as a cream solid, 118 mg, >100% yield. R.f. 0.35 (EtOAc), m.p. 178–180 °C (from hexane), LCMS: tr = 5.51 min (50% to 95% MeOH in water at 0.5 mL/min to 1.0 mL/min over 5 min), m/z M-H 323.4, HPLC: tr = 5.95 min (90% acetonitrile in H2O), 85%, 1H NMR (CDCl3, 270 MHz,): δ 3.99 (2H, s, NH2), 4.16 (1H, s, NH), 4.23 (2H, s, CH2), 6.66–6.77 (3H, m, ArH), 6.83–6.91 (4H, m, ArH), 7.07–7.16 (3H, m, ArH), 7.20–7.26 (2H, m, ArH).
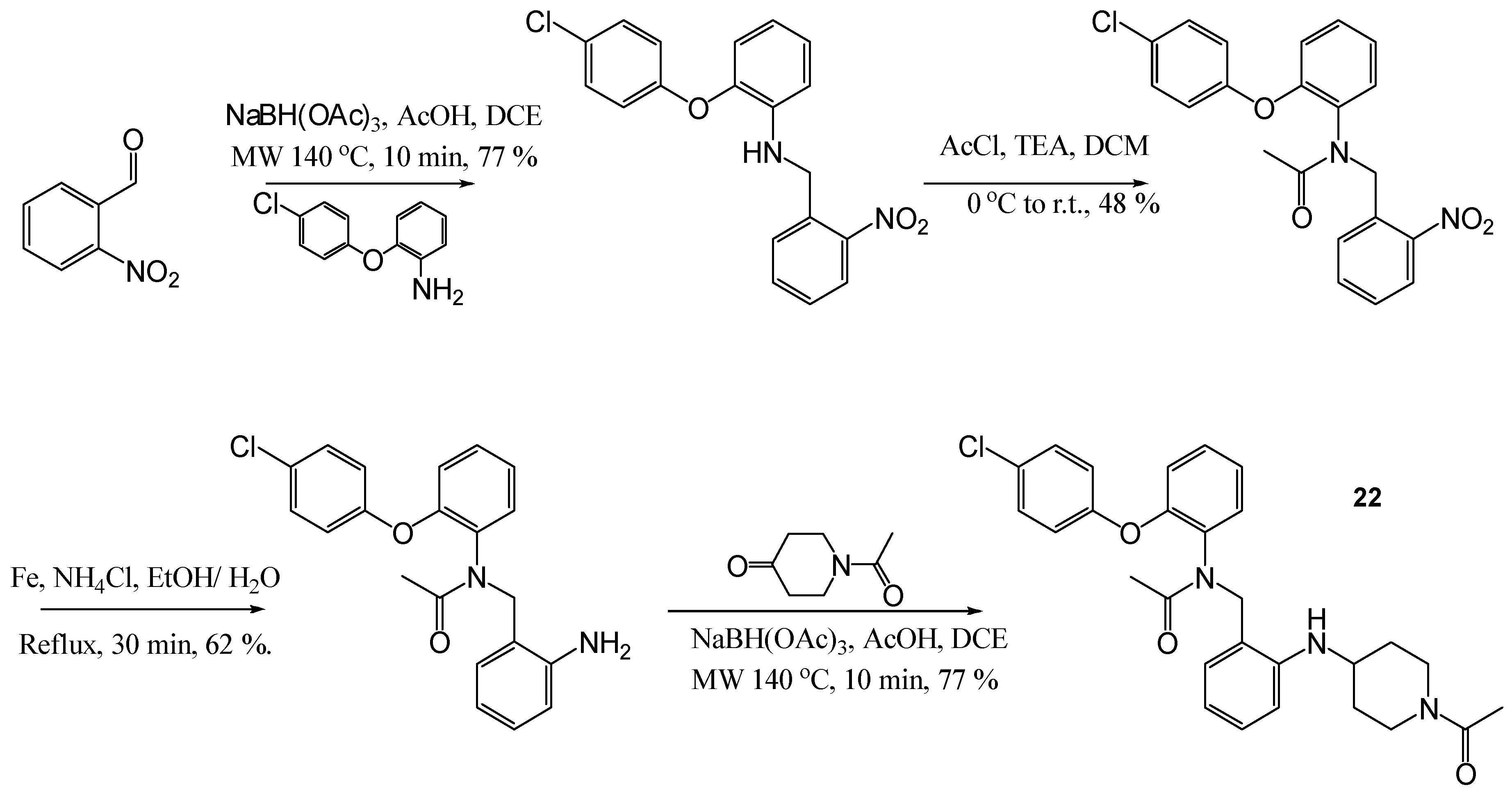
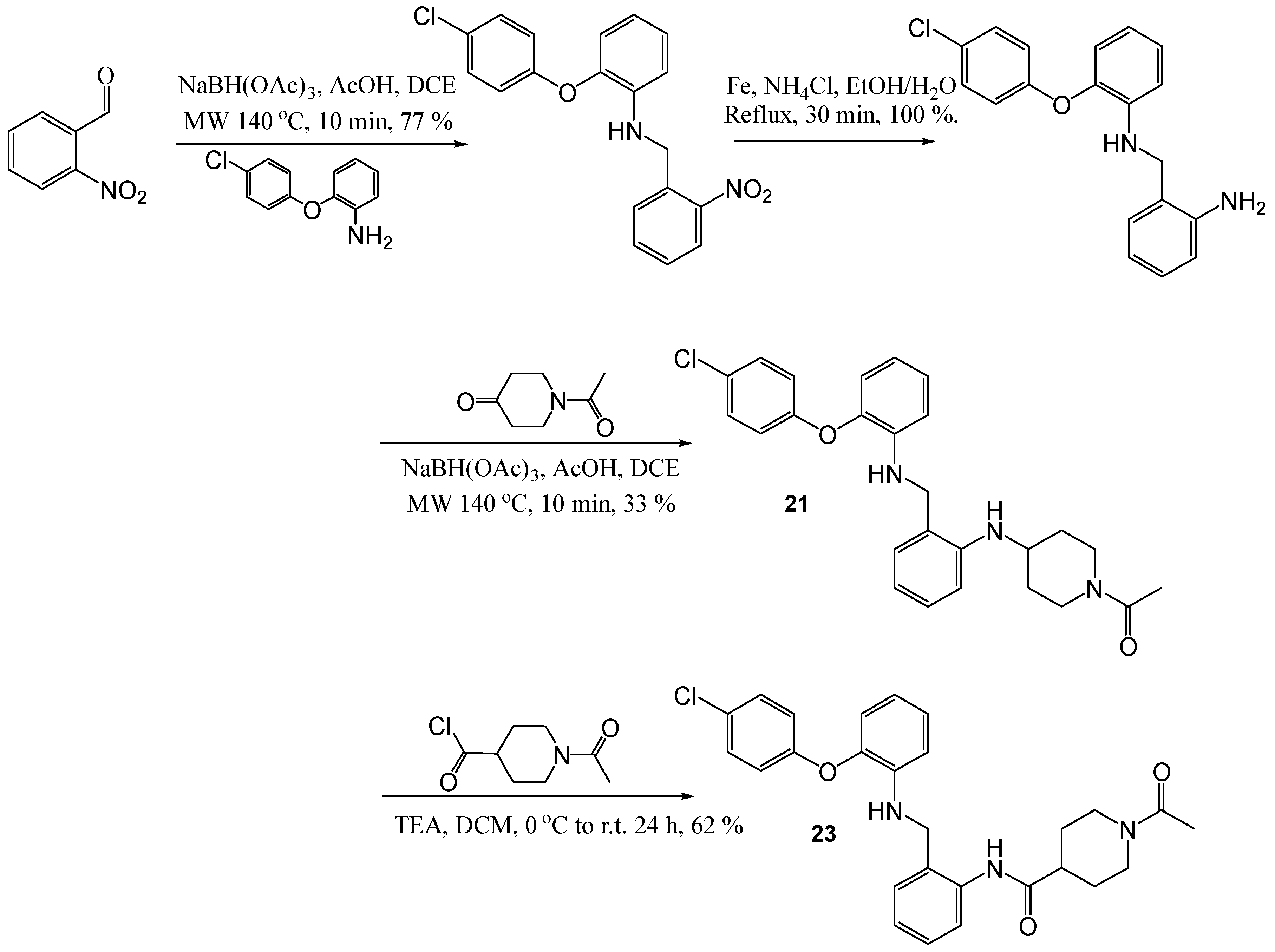
1-[4-(2-[2-(4-Chloro-phenoxy)-phenylamino]-methyl-phenylamino)-piperidin-1-yl]-ethanone (21)
To a solution of [2-(4-chloro-phenoxy)-phenyl]-(2-amino-benzyl)-amine (50 mg, 0.15 mmol) and N-benzoyl-4-piperidone (0.038 mL, 0.30 mmol) in DCE (1.5 mL), acetic acid (0.03 mL) and sodium tri-acetoxyborohydride (82 mg, 0.38 mmol) were added. The resulting reaction mixture was then subjected to microwave heating for 20 min at 140 °C. A further portion of sodium tri-acetoxy borohydride (0.45 g, 0.2 mmol) was added, and the solution was subjected to microwave heating for a further 10 min at 140 °C. NaHCO3 was added and the mixture was repeatedly extracted with DCM. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The crude mixture was purified using flash chromatography (0–10% MeOH in DCM) to afford the title compound as a cream oil, 23 mg, 33% yield. R.f. 0.2 (1:1, EtOAc: Hexane), LCMS: tr = 5.75 min (50% to 95% MeOH in water at 0.5 mL/min to 1.0 mL/min over 5 min), m/z M + Na 472.41, HPLC: tr = 6.19 min (90% acetonitrile in H2O), 96%, 1H NMR (CDCl3, 400 MHz): δ 1.25 (2H, s, CH2), 1.83–1.92 (2H, m, CH2), 2.07 (3H, s, CH3), 2.98–3.04 (1H, m, ½CH2), 3.11–3.17 (1H, m, ½CH2), 3.47–3.51 (1H, m, ½CH2), 3.56–3.62 (1H, m, ½CH2), 4.09–4.17 (1H, m, NH), 4.21 (2H, td, J = 9.6 Hz, CH2NH), 4.70 (1H, s, NH), 6.65–6.71 (2H, m, ArH), 6.75 (1H, td, J = 1.6, 7.6 Hz, ArH), 6.81–6.86 (3H, m, ArH), 6.92 (1H, dd, J = 1.2, 8.0 Hz, ArH), 7.12 (1H, td, J = 1.2, 8.0 Hz, ArH), 7.15–7.17 (1H, m, ArH), 7.19–7.23 (3H, m, ArH). 13C NMR (CDCl3, 101 MHz): δ 21.5 (CH3), 29.7, 32.1, 39.7, 44.6 (CH2), 47.5 (CH2NH), 48.7 (CH), 110.9, 112.8, 116.8, 118.4, 119.4 (ArCH), 122.1 (ArC), 125.4 (ArCH), 127.7 (ArC), 129.2, 129.6, 130.4 (ArCH), 140.2, 143.3, 145.9, 156.1 (ArC), 168.8 (CO). HRMS: Calcd. for C26H28ClN3O2 (M + H)+ 450.1943, found (M + H)+ 450.1943.
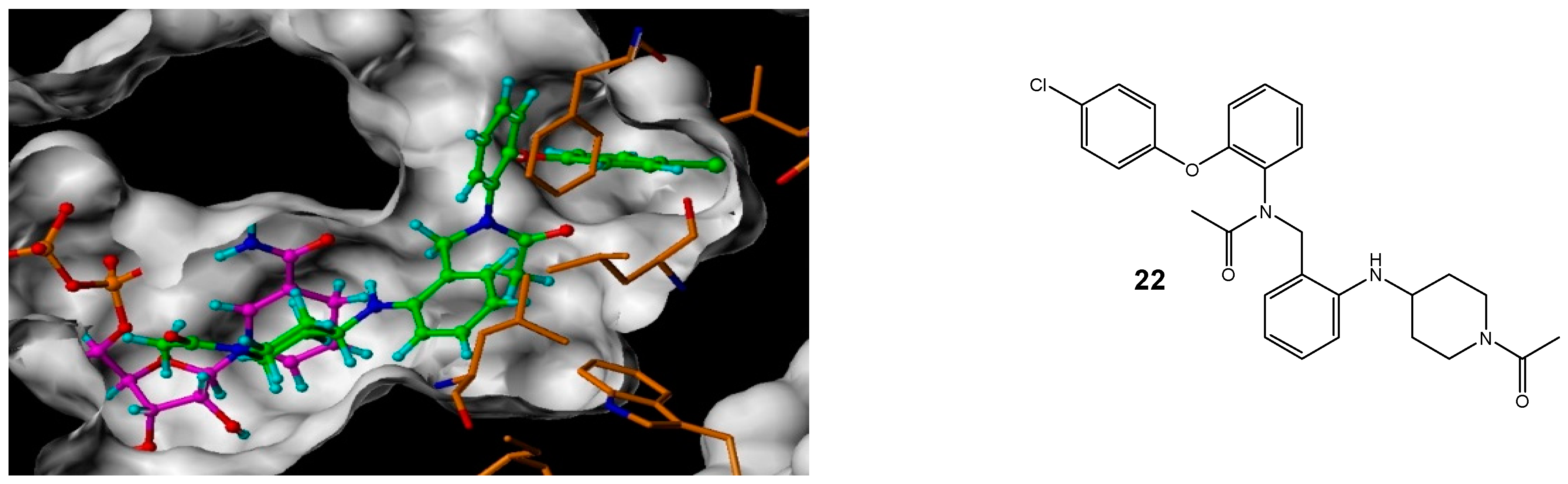
N-[2-(1-Acetyl-piperidin-4-ylamino)-benzyl]-N-[2-(4-chloro-phenoxy)-phenyl]-acetamide (22)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a cream oil, 45 mg, 63% yield. R.f. 0.72 (10% MeOH in EtOAc), LCMS: tr = 5.4 min (50% to 95% MeOH in water at 0.5 mL/min to 1.0 mL/min over 5 min), m/z M + H 492.49, HPLC: tr = 6.42 min (90% acetonitrile in H2O), 99%, 1H NMR (CDCl3, 400 MHz): δ (Multiple signals observed due to restricted rotation and, therefore, the presence of rotamers) 1.34–1.51 (1H, m, ½CH2),1.55–1.66 (1H, m, ½CH2),1.75–1.79 (1H, m, ½CH2), 1.91, 1.93 (3H, s, CH3), 1.96–2.01 (1H, m, ½CH2), 2.08, 2.09 (3H, s, CH3), 2.91–2.97 (½H, m, ¼CH2), 3.02–3.09 (½H, m, ¼CH2), 3.11–3.18 (½H, m, ¼CH2), 3.21–3.27 (½H, m, ¼CH2), 3.33–3.41 (1H, m, CH), 3.68–3.74 (½H, m, ¼CH2), 3.81–3.86 (½H, m, ¼CH2), 3.99–4.04 (½H, m, ¼CH2), 4.23–4.28 (½H, m, ¼CH2), 4.36 (½H, d, J = 14.4 Hz, ¼CH2), 4.65 (½H, d, J = 14.8 Hz, ¼CH2), 4.92 (½H, d, J = 14.4 Hz, ¼CH2), 5.25 (½H, d, J = 14.4 Hz, ¼CH2), 5.58–5.66 (1H, m, NH), 6.30–6.37 (3H, m, ArH), 6.44–6.50 (2H, m, ArH), 6.63–6.66 (½H, m, ArH), 6.72–6.74 (½H, m, ArH), 7.03–7.12 (3H, m, ArH), 7.13–7.16 (1H, m, ArH), 7.17–7.23 (2H, m, ArH). 13C NMR (CDCl3, 101 MHz): δ 21.5, 21.5, 22.0 (CH3), 30.6, 31.3, 31.9, 32.3, 39.5, 39.7, 44.7 (CH2), 48.4, 48.7 (CH), 49.5, 49.9 (CH2), 110.1, 110.2, 114.9, 115., 117.8, 118.1, 119.6, 119.7, 120.5, 120.9, 123.7, 123.8, 129.1, 129.1, 129.3, 129.4, 129.6, 129.7, 129.9 (ArCH), 131.6, 131.7 (ArC), 132.1, 132.2 (ArCH), 145.8, 145.9, 153.3, 153.8, 154.0, 154.3 (ArC), 168.7, 168.7, 171.6, 171.7 (CO). HRMS: Calcd. for C28H30ClN3O3 (M + H)+ 492.2048, found (M + H)+ 492.2049.
1-Acetyl-piperidine-4-carboxylic acid (2-[2-(4-chloro-phenoxy)-phenyl amino]-methyl-phenyl)-amide (23)
To a solution of [2-(4-chloro-phenoxy)-phenyl]- (2-amino-benzyl)-amine (48 mg, 0.15 mmol) and TEA (0.09 mL) in DCM (6 mL) at 0 °C, 1-acetylpiperidine-4-carbonyl chloride (58 mg, 0.6 mmol) was added and the resulting solution was stirred, allowed to warm to room temperature and further stirred for 24 h. NaHCO3 was added and the mixture was extracted with DCM. The organic portions were then washed with 1 M HCl. The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The title compound was obtained as an off-white solid, 44 mg, 62% yield. R.f. 0.15 (10% MeOH in DCM), m.p. 140–143 °C (from hexane), LCMS: tr = 1.25 min (95% MeOH in water), m/z M-H 476.56 HPLC: tr = 1.71 min (90% acetonitrile in H2O), 95%, 1H NMR (CDCl3, 400 MHz): δ 1.49–1.63 (2H, m, CH2), 1.74–1.83 (2H, m, CH2), 1.99 (3H, s, CH3), 2.18–2.24 (1H, m, CH), 2.49–2.57 (1H, m, CH2), 2.92–2.99 (1H, m, CH2), 3.69–3.73 (1H, m, CH2), 4.28 (3H, s, CH2NH and NH), 4.42–4.46 (1H, m, CH2), 6.77–6.86 (3H, m, ArH), 6.92–6.94 (1H, m, ArH), 7.06–7.10 (2H, m, ArH), 7.22–7.33 (5H, m, ArH), 8.05 (1H, d, J = 8.0 Hz, ArH), 8.85 (1H, s, NHCO). 13C NMR (CDCl3, 101 MHz): δ 21.4 (CH3), 28.5, 28.7, 40.8 (CH2), 43.8 (CH), 45.6, 47.8 (CH2), 113.5, 118.7, 119.2, 119.7, 122.4, 124.5, 125.3 (ArCH), 127.2, 128.2 (ArC), 128.9, 129.7, 129.8 (ArCH), 137.4, 139.3, 144.0, 155.7 (ArC), 168.7, 172.1 (CO). HRMS: Calcd. for C27H28ClN3O3 (M + Na)+ 500.1711, found (M + Na)+ 500.1705.
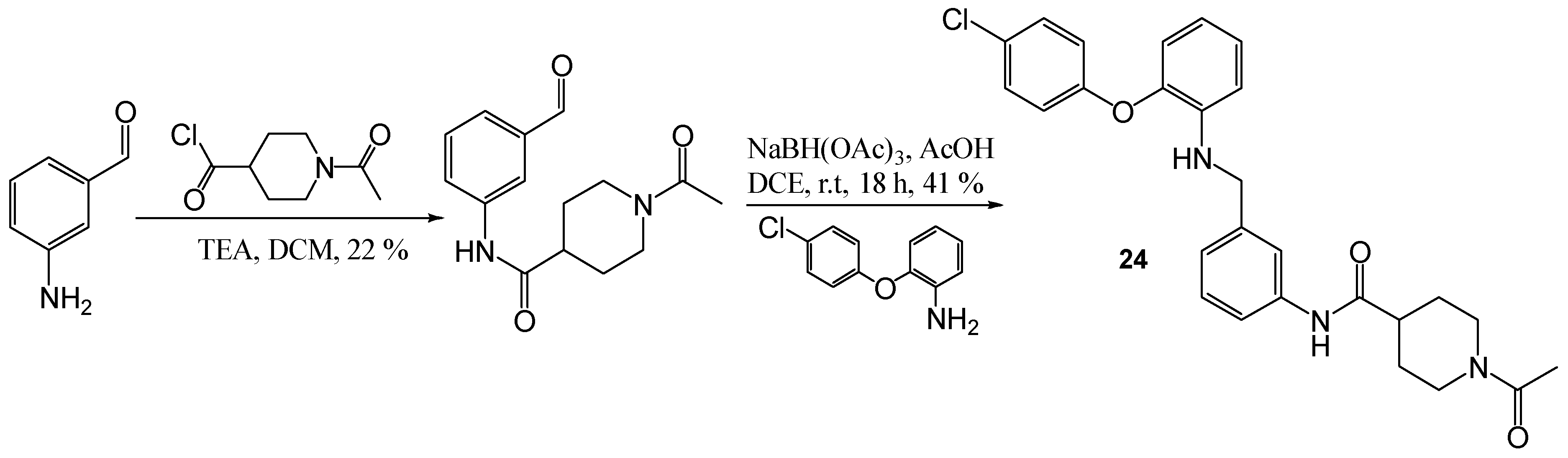
1-Acetyl-piperidine-4-carboxylic acid (3-formyl-phenyl)-amide
To a solution of 3-amino-benzaldehyde (200 mg, 1.65 mmol) and TEA (0.13 mL) in DCM (4 mL) at 0 °C, 1-acetylpiperidine-4-carbonyl chloride (0.6 g, 3.3 mmol) was added and the resulting solution was allowed to warm to room temperature and stirred for 2 days. NaHCO3 was added and the mixture was repeatedly extracted with DCM; the organic layers were then washed with HCl (1 M). The organic layers were combined and dried (MgSO4), filtered and evaporated in vacuo. The title compound was obtained as a cream oil, 97 mg, 22% yield. R.f. 0.42 (10% MeOH in DCM), LCMS: tr = 0.99 min (95% MeOH in water), m/z M-H 273.39, HPLC: tr = 1.26 min (90% acetonitrile in H2O), 81%, 1H NMR (CDCl3, 270 MHz,): δ 1.66–1.90 (2H, m, CH2), 1.95–2.03 (2H, m, CH2), 2.11 (3H, s, CH3), 2.50–2.61 (1H, m, CH), 2.66–2.76 (1H, m, ½CH2), 3.09–3.20 (1H, m, ½CH2), 3.89–3.94 (1H, m, ½CH2), 4.61–4.65 (1H, m, ½CH2), 7.48 (1H, t, J = 7.9 Hz, ArH), 7.61 (1H, td, J = 1.2, 7.7 Hz, ArH), 7.90–8.00 (4H, m, ArH and NH), 9.97 (1H, s, CHO).
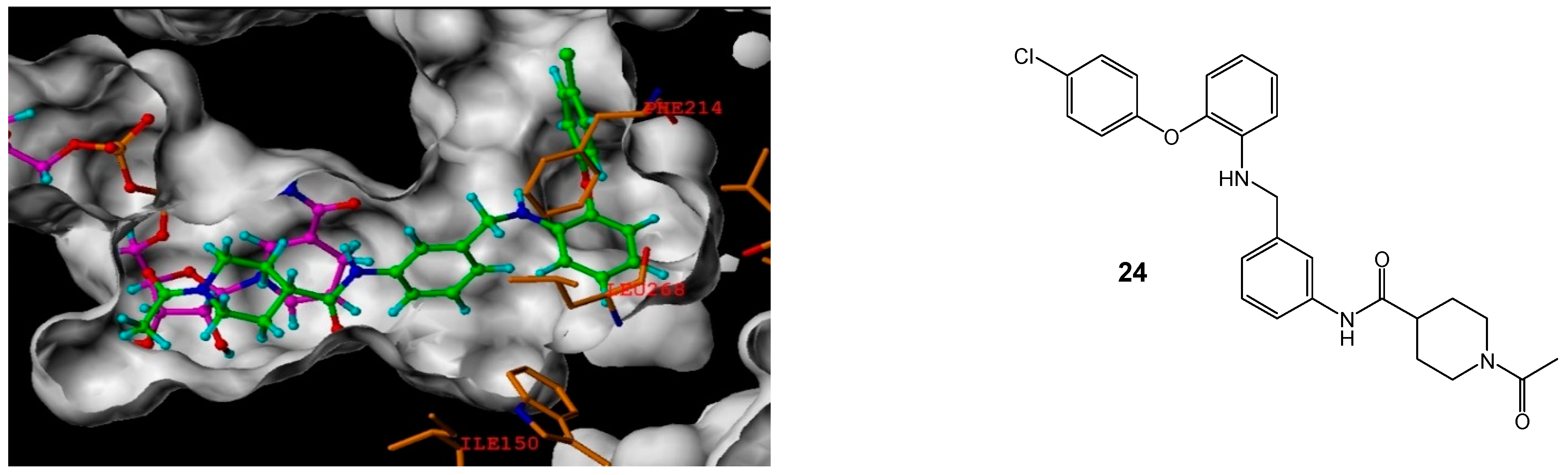
1-Acetyl-piperidine-4-carboxylic acid (3-[2-(4-chloro-phenoxy)-phenyl amino]-methyl-phenyl)-amide (24)
Using the general procedure for the reductive amination of the substituted diphenyl ether aniline with the substituted 2-acetamide benzaldehyde, the desired compound was isolated as a cream wax, 70 mg, 41% yield. R.f. 0.22 (10% MeOH in EtOAc), LCMS: tr = 5.5 min (50% to 95% MeOH in water at 0.5 mL/min to 1.0 mL/min over 5 min), m/z M-H 476.42, HPLC: tr = 1.73 min (90% acetonitrile in H2O), 93%, 1H NMR (CDCl3, 270 MHz,): δ 1.63–1.77 (2H, m, CH2), 1.78–1.90 (2H, m, CH2), 2.09 (3H, s, CH3), 2.38–2.50 (1H, m, CH), 2.63–2.73 (1H, m, ½CH2), 3.11 (1H, td, J = 2.7, 13.9 Hz, ½CH2), 3.88 (1H, d, J = 13.6 Hz, ½CH2), 4.33 (2H, s, CH2), 4.58–4.63 (2H, m, ½CH2 and NH), 6.60–6.66 (2H, M, ArH), 6.82 (1H, dd, J = 1.5, 8.4 Hz, ArH), 6.86–6.92 (2H, m, ArH), 6.95–7.06 (2H, m, ArH), 7.21–7.28 (3H, m, ArH), 7.39–7.44 (3H, m, ArH and NH). 13C NMR (CDCl3, 68 MHz): δ 21.6 (CH3), 28.6, 28.9, 41.0, (CH2), 44.1 (CH), 45.8, 47.6 (CH2), 112.0, 117.2, 118.5, 118.7, 118.8, 119.4, 123.2, 125.4 (ArCH), 127.7 (ArC), 129.4, 129.8 (ArCH), 129.8, 138.1, 140.2, 140.5 (ArC), 169.0, 172.3 (CO). HRMS: Calcd. for C27H28ClN3O3 (M + H)+ 478.1892, found (M + H)+ 478.1878.
N-(2-Acetyl-phenyl)-acetamide
A solution of 2-aminoacetopheone (2.0 g, 14.8 mmol) in DCM (80 mL) was cooled to 0 °C, and TEA (2.4 mL) and acetyl chloride (2.06 mL, 30 mmol) were added. The resulting solution was stirred at r.t. for 30 min. NaHCO3 was added and the solution was extracted; the organic layers were then washed with HCl (1M) and brine. The organic layers were dried (MgSO4), filtered and evaporated in vacuo to yield the desired product as a brown solid, 2.3 g, 89% yield. R.f. 0.49 (EtOAc), m.p. 68–70 °C, LCMS: tr = 1.32 min (80% MeOH in water), m/z M-H 175.79, HPLC: tr = 1.60 min (90% acetonitrile in H2O), 94%, 1H NMR (CDCl3, 270 MHz): δ 2.15 (3H, s, CH3), 2.58, (3H, s, CH3), 6.99–7.06 (1H, m, ArH), 7.43–7.49 (1H, m, ArH), 7.80 (1H, dd, J = 1.5, 8.15 Hz, ArH), 8.65 (1H, dd, J = 1.0, 8.4 Hz, ArH), 11.63 (1H, br.s, NH). 13C NMR (CDCl3, 68 MHz): δ 25.7, 28.8 (CH3), 120.8 (ArCH), 121.7 (ArC), 122.4, 131.7, 135.3 (ArCH), 141.1 (ArC), 169.6, 202.9 (CO). HRMS: Calcd. for C10H11NO2 (M + H)+ 178.0863, found (M + H)+ 178.0858.
N-(2–1-[2-(4-Chloro-phenoxy)-phenylamino]-ethyl-phenyl)-ethylamine (25)
A solution of 2-(4-chloro-phenoxy)-phenylamine (298 mg, 1.4 mmol), N-(2-acetyl-phenyl)-acetamide, (200 mg, 1.13 mmol) and chloro-tri-isopropoxy-titanium IV (0.53 mL, 2.26 mmol) in toluene (15 mL) was stirred at ambient temperature for 4 days. NaHCO3 was added and the mixture was extracted with EtOAc, dried (MgSO4) and evaporated to dryness. The residue was redissolved in THF (20 mL) and cooled to 0 °C, succinic acid (270 mg, 2.26 mmol) and borane (1M in THF, 2.3 mL, 2.26 mmol) were added. The reaction was slowly warmed to r.t. and stirred for 8h. NaHCO3 was added, and the volatile solvents removed in vacuo; the mixture was then extracted with EtOAc and dried (MgSO4). The crude material was purified by flash chromatography (0–100% DCM in hexane) to yield the product as an oil, 79 mg, 19% yield. LCMS: tr = 1.42 min (95% MeOH in water), m/z M-H 365.33, HPLC: tr = 4.49 min (90% acetonitrile in water), 97%, 1H NMR (CDCl3, 270 MHz,): δ 1.17 (3H, t, J = 7.2 Hz, CH3CH2), 1.56 (3H, d, J = 6.7 Hz, CH3CH), 3.10 (2H, q, J = 14.1 Hz, CH2), 4.22 (1H, d, J = 6.0 Hz, NH), 4.53 (1H, q, J = 13.3 Hz, CH), 4.59 (1H, br.s, NH), 6.66–6.93 (7H, m, ArH), 7.01 (1H, td, J = 7.9, 1.5 Hz, ArH), 7.16–7.31 (4H, m, ArH). 13C NMR (CDCl3, 68 MHz): 14.9, 19.9 (CH3), 38.1 (CH2), 50.9 (CH), 111.1, 113.6, 117.0, 118.0, 118.6, 119.5, 125.5, 126.5 (ArCH), 128.6, 127.8 (ArC), 128.3, 129.7 (ArCH), 13.7, 143.3, 146.7, 156.4 (ArC). HRMS: Calcd. for C22H23ClN2O (M + Na)+ 389.1386, found (M + Na)+ 389.1391.
N-(2-(1-[2-(4-Chloro-phenoxy)-phenylamino]-but-2-enyl)-phenyl)-acetamide (26)
N-(2-([2-(4-Chloro-phenoxy)-phenylimino]-methyl)-phenyl)-acetamide (500 mg, assumed 100% pure, 1.4 mmol) was dissolved in THF (15 mL) and cooled to 0 °C under a N2 atmosphere, BF3OEt2 (0.18 mL, 1.4 mmol) and allyl magnesium bromide (1 M in ether, 4.2 mL, 4.2 mmol) were added. The resulting solution was stirred at r.t. for 18 h. The reaction was then quenched with sat. NH4Cl solution then extracted with EtOAc and dried (MgSO4). The crude product was purified by flash chromatography (0–50% EtOAc in hexane) to yield the desired product as a light brown solid, 320 mg, 57% yield. R.f. 0.36 (DCM), LCMS: tr = 1.59 min (95% MeOH in water), m/z M + H (+Na) 429.13, M + H 407.15, HPLC: tr = 2.45 min (90% acetonitrile in water), 92%, HRMS: Calcd. for C24H23ClN2O2 (M + H)+ 407.1521, found (M + H)+ 407.1503. 1H NMR (CDCl3, 400 MHz,): δ 1.86 (3H, s, CH3), 2.54–2.62 (2H, m, CH2), 4.31 (1H, t, J = 6.4 Hz, CHNH), 4.60 (1H, s, NH), 5.10 (1H, s, ½CH2CH), 5.13 (1H, d, J = 5.2 Hz, ½CH2CH), 5.65–5.75 (1H, m, CHCH2), 6.66 (1H, d, J = 8.0 Hz, ArH), 6.77 (1H, t, J = 8.0 Hz, ArH), 6.87–6.96 (4H, m, ArH), 7.13 (1H, t, J = 7.6 Hz, ArH), 7.26–7.31 (4H, m, ArH), 8.05 (1H, d, J = 8.4 Hz, ArH), 9.40 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): 24.3 (CH3), 40.6 (CH), 58.9 (CH2), 114.9, 118.0 (ArCH), 119.3 (CH2), 119.8, 120.0, 123.1, 124.7, 125.6 (ArCH), 128.0 (ArC), 128.2, 128.2, 129.8 (ArCH), 130.7 (ArC), 134.0 (CH), 136.9, 139.3, 143.5, 156.1 (ArC), 168.1 (CO).
N-(2-(1-[2-(4-Chloro-phenoxy)-phenylamino]-2-phenyl-ethyl)-phenyl)-acetamide (27)
N-(2-([2-(4-Chloro-phenoxy)-phenylimino]-methyl)-phenyl)-acetamide (111 mg, assumed 100% pure, 0.3 mmol) was dissolved in THF (5 mL) and cooled to 0 °C under a N2 atmosphere, BF3OEt2 (0.04 mL, 0.3 mmol) and benzyl magnesium bromide (2 M in THF, 0.3 mL, 1.2 mmol) were added. The resulting solution was stirred at r.t. for 18 h. The reaction was then quenched with sat. NH4Cl solution, extracted with EtOAc and dried (MgSO4). The crude product was purified by flash chromatography (0–20% EtOAc in DCM) to yield the desired product as an oil, 20 mg, 14% yield. R.f. 0.25 (DCM), LCMS: tr = 1.26 min (95% MeOH in water), m/z M-H 455.15, HPLC: tr = 2.73 min (90% acetonitrile in water), 97%, 1H NMR (CDCl3, 400 MHz,): δ 1.82 (3H, s, CH3), 3.02–3.04 (2H, m, CH2), 4.43 (1H, t, J = 8.0 Hz, CH), 4.50 (1H, br.s, NH), 6.52 (1H, d, J = 6.8 Hz, ArH), 6.63–6.74 (4H, m, ArH), 6.79–6.84 (1H, m, ArH), 6.98–7.00 (1H, m, ArH), 7.05 (1H, t, J = 7.2 Hz, ArH), 7.15–7.24 (7H, m, ArH), 7.91 (1H, d, J = 8.4 Hz, ArH), 8.90 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): δ 24.3 (CH3), 42.9 (CH2), 60.8 (CH), 114.9, 117.9, 119.6, 119.7, 123.4, 124.9, 125.6, 127.2 (ArCH), 127.9 (ArC), 128.1, 128.2, 128.9, 129.1, 129.8 (ArCH), 131.2, 136.7, 136.8, 139.3, 143.4, 156.0 (ArC), 168.2 (CO). HRMS: Calcd. for C28H25ClN2O2 (M + H)+ 457.1677, found (M + H)+ 457.1666.
N-(2-(1-[2-(4-Chloro-phenoxy)-phenylamino]-butyl)-phenyl)-acetamide (28)
To a solution of N-(2-(1-[2-(4-chloro-phenoxy)-phenylamino]-but-2-enyl)-phenyl)-acetamide, (50 mg, 0.12 mmol) in EtOAc (25 mL) had Pd/C (15 mg) added. The solution was then stirred under a H2 atmosphere for 15 min and filtered through Celite. Purification by flash chromatography (0–50% EtOAc in hexane) afforded the desired product, 44 mg, 88% yield. R.f. 0.42 (EtOAc), LCMS: tr = 3.72 min (90% MeOH in water), m/z M + H 409.00, HPLC: tr = 4.69 min (90% MeOH in water), 99%, 1H NMR (CDCl3, 400 MHz,): δ 0.90 (3H, t, J = 7.6 Hz, CH2CH3), 1.21–1.39 (2H, m, CH2), 1.79–1.86 (2H, m, CH2), 1.87 (3H, s, CH3CO), 4.29 (1H, t, J = 7.6 Hz, CH), 4.37 (1H, br.s, NH), 6.70 (1H, d, J = 8.0 Hz, ArH), 6.73–6.78 (1H, m, ArH), 6.85–6.87 (1H, m, ArH), 6.90–6.96 (3H, m, ArH), 7.11 (1H, t, J = 7.2 Hz, ArH), 7.25–7.31 (4H, m, ArH), 8.05 (1H, d, J = 8.0 Hz, ArH), 9.36 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): 13.7 (CH3CH2), 19.6 (CH2), 24.4 (CH3CO), 38.0 (CH2), 60.0 (CH), 114.7, 118.3, 119.4, 119.5, 123.0, 124.4, 125.4, 127.9, 128.1, 128.5, 129.9 (ArCH), 130.9, 136.8, 139.3, 143.7, 156.0 (ArC), 168.1 (CO). HRMS: Calcd. for C24H25ClN2O2 (M + H)+ 409.1677, found (M + H)+ 409.1677.
N-(2-(1-[2-(4-Chloro-phenoxy)-phenylamino]-ethyl)-phenyl)-acetamide (29)
A cerium chloride suspension was prepared. CeCl3.7H2O (stored in the oven, 515 mg, 1.38 mmol) was heated under high vacuum for 15 min and allowed to cool to r.t., and then to 0 °C in an ice bath. To this THF (3 mL) and methyl magnesium bromide (3 M in diethyl ether, 0.46 mL, 1.38 mmol) were added, this was then stirred at r.t. for 2 h. To this N-(2-([2-(4-chloro-phenoxy)-phenylimino]-methyl)-phenyl)-acetamide intermediate (166 mg, 0.46 mmol) was added and stirred at r.t. for a further 18 h. NaHCO3 was added and the mixture was extracted with EtOAc, dried (MgSO4) and purified by flash chromatography (0–100% DCM) to yield the desired product 14 mg, 8% yield. R.f. 0.56 (DCM with TEA), LCMS: tr = 1.08 min (90% MeOH in water), m/z M + Na 403.20, HPLC: tr = 2.6 min (90% acetonitrile in water), 94%, 1H NMR (CDCl3, 270 MHz,): δ 1.55 (3H, d, J = 6.6 Hz, CH3CH), 1.90 (3H, s, CH3CO), 4.25 (1H, d, J = 3.0 Hz, CHNH), 4.52–4.54 (1H, m, CH), 6.73–6.78 (2H, m, ArH), 6.84–6.99 (4H, m, ArH), 7.11 (1H, t, J = 7.7 Hz, ArH), 7.24–7.31 (4H, m, ArH), 8.02 (1H, d, J = 8.0 Hz, ArH), 9.16 (1H, br.s, NH). 13C NMR (CDCl3, 101 MHz): 21.5, 24.4 (CH3), 53.9 (CH), 114.6, 118.5, 119.4, 119.6, 123.2, 124.7, 125.4, 127.3, 128.1 (ArCH), 128.2 (ArC), 129.9 (ArCH), 132.0, 136.8, 138.9, 143.9, 155.9 (ArC), 168.2 (CO). HRMS: Calcd. for C22H21ClN2O2 (M + H)+ 381.1364, found (M + H)+ 381.1352.
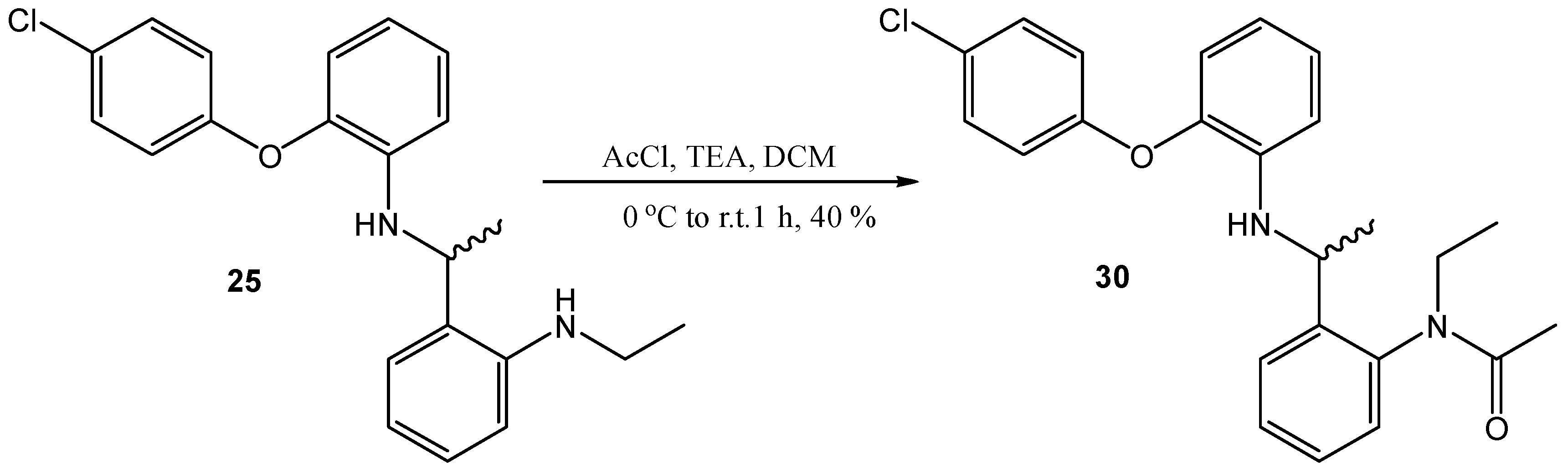
N-(4-(1-[2-(4-Chloro-phenoxy)-phenylamino]-ethyl)-phenyl)-N-ethyl-acetamide (30)
N-(4-(1-[2-(4-Chloro-phenoxy)-phenylamino]-ethyl)-phenyl)-ethane (50 mg, 0.14 mmol) was dissolved in DCM (1 mL) and cooled to 0°C, acetyl chloride (0.04 mL, 0.56 mmol) and TEA (0.02 mL, 0.42 mmol) were added. This was then allowed to warm to room temperature and stirred for 1h. Saturated NaHCO3 solution was added, and the mixture was extracted with DCM, dried (MgSO4) and purified by flash chromatography to yield the title compound as an off-white oil, 23 mg, 40% yield. R.f. 0.55 (EtOAc), LCMS: tr = 1.37 min (95% MeOH in water), m/z M-H 407.34, HPLC: tr = 3.03 min (90% acetonitrile in H2O), 96%, 1H NMR (CDCl3, 270 MHz,): δ 1.15 (3H, dt, J = 6.8, 10.4 Hz, CH3CH2), 1.43 (3H, dd, J = 6.4, 15.6 Hz, CH3CH), 1.74 (3H, d, J = 25.6 Hz, CH3CO), 3.07–3.15 (1H, m, ½CH2), 4.24–4.36 (2H, m, ½CH2 and NH), 4.64–4.75 (1H, m, CH), 6.51 (1H, dd, J = 1.6, 8.4 Hz, ArH), 6.57–6.67 (2H, m, ArH), 6.74–6.84 (2H, m, ArH), 6.86–6.90 (1H, m, ArH), 6.92–6.97 (1H, m, ArH), 7.06 (1H, td, J = 1.6, 8.0 Hz, ArH), 7.21–7.35 (4H, m, ArH), 7.39–7.47 (1H, m, ArH). 13C NMR (CDCl3, 68 MHz): 12.8, 22.5, 23.1 (CH3), 43.5 (CH2), 47.5 (CH), 112.7, 117.7, 118.8, 119.4, 125.3, 126.9, 128.3, 129.4, 129.7, 130.3 (ArCH), 138.9, 139.9, 141.7, 142.4, 143.1, 156.3 (ArC), 170.5 (CO). HRMS: Calcd. for C24H25ClN2O2 (M + Na)+ 431.1497, found (M + Na)+ 431.1487.
1-Bromo-2-phenoxy-benzene
A mixture of 2-bromophenol (0.211 mL, 2 mmol), phenylboronic acid (490 mg, 4 mmol), copper acetate (364 mg, 2 mmol), TEA (1.38 mL, 10 mmol) and 4Å molecular sieves in DCM (25 mL) was stirred at r.t. for 18 h. The slurry was filtered through Celite and concentrated in vacuo. This was then diluted with EtOAc and NaHCO3 solution, extracted and the organic portions were washed with brine and dried (MgSO4). The crude mixture was purified by flash chromatography (hexane) to yield the desired product as a colourless oil, 232 mg, 47% yield. R.f. 0.75 (DCM), LCMS: tr = 1.3 min (95% MeOH in water), m/z M-H 246.84, 248.86, HPLC: tr = 2.88 min (90% acetonitrile in water), 98%, 1H NMR (CDCl3, 270 MHz,): δ 6.95–7.04 (4H, m, ArH), 7.11 (1H, td, J = 1.1, 8.0 Hz, ArH), 7.22–7.37 (3H, m, ArH), 7.61–7.65 (1H, m, ArH). 13C NMR (CDCl3, 68 MHz): 115.0 (ArC), 118.2, 120.7, 123.5, 125.1, 128.8, 129.9, 133.9 (ArCH), 153.8, 156.9 (ArC).
1-Bromo-2-phenoxy-4′-chlorobenzene
A mixture of 2-bromophenol (0.2 mL, 2 mmol), phenylboronic acid (600 mg, 4 mmol), copper acetate (350 mg, 2 mmol), TEA (1.4 mL, 10 mmol) and powdered 4 Å molecular sieves (~2 g) in DCM (25 mL) was stirred at r.t. for 18 h. The slurry was filtered through Celite and concentrated in vacuo. This was then diluted with EtOAc and NaHCO3 solution, extracted and the organic portions were washed with brine and dried (MgSO4). The crude mixture was purified by flash chromatography (hexane) to yield the desired product as a colourless oil, 320 mg, 59% yield. R.f. 0.72 (DCM), HPLC: tr = 3.29 min (90% acetonitrile in water), >99%, 1H NMR (CDCl3, 270 MHz,): δ 6.85–6.91 (1H, m, ArH), 6.96 (1H, dd, J = 1.4, 8.0 Hz, ArH), 7.00–7.07 (1H, m, ArH), 7.25–7.29 (3H, m, ArH), 7.62 (1H, dd, J = 1.6, 8.0 Hz, ArH).
1-(2-Nitro-phenyl)-ethanone-O-methyl-oxime
To a solution of 2-nitroacetophenone (1.9 g, 10.9 mmol), methoxyamine hydrochloride (0.96 g, 10.9 mmol) in anhydrous pyridine (38 mL) and anhydrous EtOH (38 mL), powdered 4Å molecular sieves (~1 g) were added. The resulting mixture was heated at reflux for 3 h. The resulting mixture was filtered through Celite to remove the molecular sieves and then evaporated to dryness. The solid was redissolved in EtOAc and extracted with 20% NaHCO3 solution; this was then dried (MgSO4) and evaporated in vacuo to yield the desired compound as a mixture of enantiomers, yellow oil, 1.95 g, 87% yield. The product was used crude in following reactions. R.f. 0.55 (DCM), HPLC: tr = 1.87 min, 58%, tr = 2.39 min, 29% (90% acetonitrile in water), LCMS: tr = 3.30 min, m/z M + H 195.4, tr = 4.00 min, m/z M + H 195.3 (70% MeOH in water). 1H NMR (CDCl3, 400 MHz,): Major isomer: δ 2.54 (3H, s, CH3), 3.69 (3H, s, OCH3), 7.23–8.08 (4H, m, ArH). Minor isomer: δ 2.14 (3H, s, CH3), 3.94 (3H, s, OCH3), 7.23–8.08 (4H, m, ArH).
1-(2-Nitro-phenyl)-ethylamine hydrochloride
A solution of 1-(2-nitro-phenyl)-ethanone-O-methyl-oxime (1.95 g, 10.05 mmol) in THF (7 mL) was cooled to 0 °C, borane/ THF complex (28 mL, 28.1 mmol) was added and the resulting solution was then heated at reflux for 6 h. The reaction was then cooled to −20 °C and water (2 mL) was added slowly followed by aq. 20% KOH solution (2 mL) over 20 min. The resulting mixture was then heated at reflux for a further 2 h and then poured into DCM. The mixture was then extracted with brine and dried (MgSO4). To form the salt, the product was redissolved in DCM and then concentrated HCl (1.5 mL) was added to the mixture and stirred for 1 h. The resulting solid was removed by filtration and washed with ether and dried, 345 mg, 17% yield. R.f. 0.32 (Hexane: DCM, 1:1), LCMS: tr = 1.33 min (70% MeOH in water), m/z M + H 167.2 (free base), HPLC: tr = 2.09 min (90% acetonitrile in water), >99%, 1H NMR (CDCl3, 400 MHz,): δ 1.60 (3H, d, J = 6.8 Hz, CH3), 4.77–4.80 (1H, m, CH), 7.63–7.67 (1H, m, ArH), 7.86 (1H, td, J = 1.2, 7.6 Hz, ArH), 8.03–8.05 (2H, m, ArH), 8.72 (2H, br, s, NH2).
(2-Phenoxy-phenyl)-(1-phenyl-ethyl)-amine
Palladium acetate (19 mg, 10 mol%) and rac-BINAP (51 mg, 10 mol%) were placed into an oven-dried flask; this was then evacuated and backfilled with N2. To this, 1-phenylethylamine (100 mg, 0.83 mmol), 1-bromo-2-phenoxy-benzene (185 mg, 0.74 mmol) and toluene (1 mL) were then added (via syringe). This was stirred for 10 min at r.t. Sodium t-butoxide (95 mg, 1 mmol) and further toluene (1 mL) were then added. The resulting solution was heated to reflux for 18 h. The mixture was then filtered through Celite and purified by flash chromatography (0–50% DCM in hexane) to yield the desired product as a pale cream oil, 104 mg, 49% yield. R.f. 0.45 (1:1, DCM: Hexane), LCMS: tr = 2.18 min (90% MeOH in water), m/z M + H 290.10, HPLC: tr = 3.11 min (90% acetonitrile in water), 98%, 1H NMR (CDCl3, 270 MHz,): δ 1.47 (3H, d, J = 6.3 Hz, CH3), 4.45–4.58 (2H, m, CH and NH), 6.46 (1H, dd, J = 1.1, 8.0 Hz, ArH), 6.56 (1H, td, J = 1.4, 7.9 Hz, ArH), 6.80–6.89 (2H, m, ArH), 6.99–7.11 (3H, m, ArH), 7.18–7.36 (7H, m, ArH). 13C NMR (CDCl3, 68 MHz): 25.2 (CH3), 53.3 (CH), 112.7, 116.7, 117.6, 119.2, 122.8, 124.8, 125.9, 126.9, 128.7, 129.8 (ArCH), 139.6, 143.0, 145.2, 157.7 (ArC). HRMS: Calcd. for C20H19NO (M + H)+ 290.1539, found (M + H)+ 290.1529.
[2-(4-Chloro-phenoxy)-phenyl]-(1-phenyl-ethyl)-amine
Palladium acetate (10 mg, 10 mol%) and rac-BINAP (26 mg, 10 mol%) were placed into an oven-dried flask, which was evacuated and backfilled with N2. To this was then added (via syringe) 1-phenylethylamine (50 mg, 0.42 mmol), 1-bromo-2-phenoxy-4′chlorobenzene (105 mg, 0.38 mmol) and toluene (1 mL). This was stirred for 10min at r.t. Sodium t-butoxide (50 mg, 0.5 mmol) and a further portion of toluene (1 mL) were then added. The resulting solution was heated to reflux for 3 h. This was then filtered through Celite and purified by flash chromatography (hexane) to yield the desired product as a white solid, 51 mg, 41% yield. R.f. 0.35 (hexane), m.p. 84–86 °C, LCMS: tr = 4.67 min (90% MeOH in water), m/z M-H 322.3, HPLC: tr = 4.30 min (90% acetonitrile in water), 97%, 1H NMR (CDCl3, 270 MHz,): δ 1.48 (3H, d, J = 6.0 Hz, CH3), 4.49–4.53 (2H, m, CH and NH), 6.48 (1H, t, J = 8.0 Hz, ArH), 6.92–6.95 (2H, m, ArH), 7.20–7.31 (7H, m, ArH). 13C NMR (CDCl3, 68 MHz): 25.2 (CH3), 53.3 (CH), 113.0, 116.9, 118.8, 119.3, 125.3, 125.9, 127.1 (ArCH), 127.8 (ArC), 128.8, 129.8 (ArCH), 139.5, 142.6, 145.1, 156.4 (ArC). HRMS: Calcd. for C20H18ClNO (M + H)+ 324.1150, found (M + H)+ 324.1136.
[2-(4-Chloro-phenoxy)-phenyl]-[1-(2-nitro-phenyl)-ethyl]-amine
Palladium acetate (15 mg, 10 mol%), rac-BINAP (45 mg, 10 mol%) and 1-(2-nitro-phenyl)-ethylamine hydrochloride (151 mg, 0.75 mmol) were placed into an oven-dried flask, which was evacuated and backfilled with N2. To this, 1-bromo-2-phenoxy-4′chlorobenzene (190 mg, 0.68 mmol) and toluene (2 mL) were then added (via syringe). This was stirred for 10 min at r.t. Sodium t-butoxide (195 mg, 2.04 mmol) and a further portion of toluene (2 mL) were then added. The resulting solution was heated to reflux for 24 h. The slurry was then filtered through Celite and purified by flash chromatography (0–100% DCM in hexane) to yield the desired product as a yellow oil, 120 mg, 48% yield. R.f. 0.45 (1:1, Hexane: DCM), LCMS: tr = 3.85 min (90% MeOH in water), m/z M-H 367.50, 1H NMR (CDCl3, 400 MHz,): δ 1.51 (3H, d, J = 6.8 Hz, CH3), 5.16 (1H, q, J = 6.4 Hz, CH), 6.28 (1H, dd, J = 1.2, 7.6 Hz, ArH), 6.56 (1H, td, J = 1.2, 7.6 Hz, ArH), 6.76 (1H, dd, J = 1.6, 8.4 Hz, ArH), 6.82 (1H, td, J = 1.2, 7.2 Hz, ArH), 6.89–6.93 (2H, m, ArH), 7.24–7.28 (2H, m, ArH), 7.31–7.35 (1H, m, ArH), 7.47 (1H, td, J = 1.2, 7.6 Hz, ArH), 7.54 (1H, dd, J = 1.2, 8.0 Hz, ArH), 7.88 (1H, dd, J = 1.2, 8.4 Hz, ArH). 13C NMR (CDCl3, 101 MHz): δ 24.1 (CH3), 48.7 (CH), 112.3, 117.5, 118.7, 119.1, 124.7, 125.2, 127.3, 127.8 (ArCH), 127.9 (ArC), 129.7, 133.6 (ArCH), 138.4, 140.4, 142.6, 148.7, 156.1 (ArC).
[2-(4-Chloro-phenoxy)-phenyl]-[1-(2-amino-phenyl)-ethyl]-amine
Using the general procedure for the reduction of the substituted 2-nitrobenzylalcohol, but with a shortened reaction time of 10min at reflux, the product was isolated as a yellow oil, 12 mg, 25% yield. R.f. 0.32 (DCM), LCMS: tr = 2.81 min (90% MeOH in water), m/z M-H 337.60, 1H NMR (CDCl3, 400 MHz,): δ 1.53 (3H, d, J = 6.8 Hz, CH3), 4.06 (2H, br.s, NH2), 4.21 (1H, br.s, NH), 4.54–4.56 (1H, m, CH), 6.63–6.67 (2H, m, ArH), 6.70–6.77 (2H, m, ArH), 6.81 (1H, dd, J = 0.8, 7.6 Hz, ArH), 6.87–6.91 (2H, m, ArH), 6.95–6.99 (1H, m, ArH), 7.07 (1H, td, J = 0.8, 7.2 Hz, ArH), 7.19 (1H, dd, J = 1.2, 7.6 Hz, ArH), 7.23–7.27 (2H, m, ArH). 13C NMR (C(ArC), 128.0, 129.6 (ArCH), 139.5, 143.1, 144.8, 156.2 (ArC).
N-[2-(1-Acetylpiperidin-4-ylamino)benzyl]-N-[2-(4-chlorophenoxy)phenyl]acetamide (30)
To a solution of [2-(4-chloro-phenoxy)-phenyl]-[1-(2-amino-phenyl)-ethyl]-amine (20 mg, 0.06 mmol) and TEA (0.008 mL) in DCM (1 mL) at 0 °C, acetyl chloride (0.009 mL) was added and the resulting solution was warmed to r.t. and stirred for 1 h. NaHCO3 was added and the mixture extracted with DCM; the organic layers were then washed with 1M HCl. The organic layers were combined and dried (MgSO4) and evaporated in vacuo. Purification by flash chromatography (0–10% MeOH in DCM) afforded the desired product as a pale cream oil, 22 mg, 96% yield.
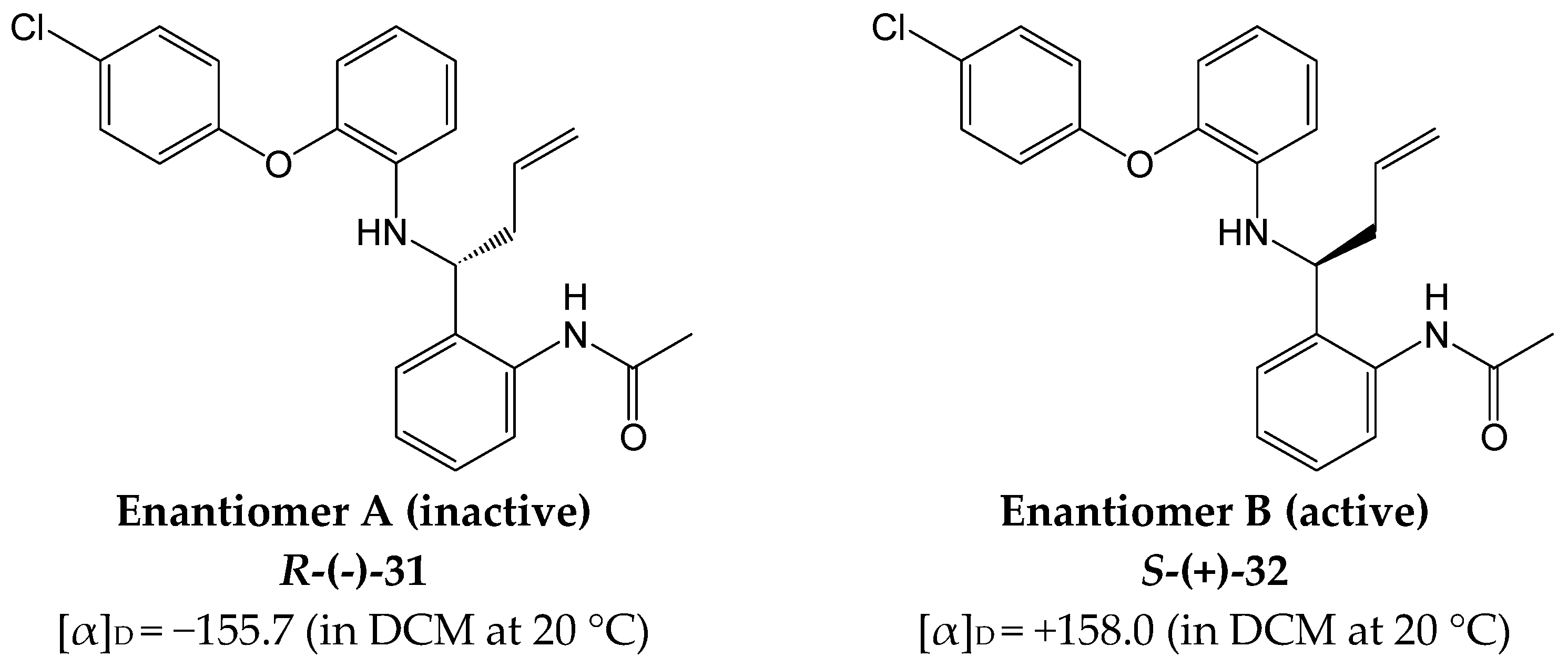
R-(-)-N-(2-(1-[2-(4-Chloro-phenoxy)-phenylamino]-but-2-enyl)-phenyl)-acetamide (31)
m.p. 139–140 °C (from hexane/DCM), LCMS (Chiracel AD-H column): tr = 11.5 min (80% MeOH in water), m/z M-H 405.2, HPLC (Chiracel AD-H column): tr = 9.00 min (80% MeOH in water), >99%, [α]D = −155.7, 1H NMR (CDCl3, 400 MHz,): δ 1.86 (3H, s, CH3), 2.54–2.62 (2H, m, CH2), 4.31 (1H, t, J = 6.4 Hz, CHNH), 4.60 (1H, s, NH), 5.10 (1H, s, ½CH2CH), 5.13 (1H, d, J = 5.2 Hz, ½CH2CH), 5.65–5.75 (1H, m, CHCH2), 6.66 (1H, d, J = 8.0 Hz, ArH), 6.77 (1H, t, J = 8.0 Hz, ArH), 6.87–6.96 (4H, m, ArH), 7.13 (1H, t, J = 7.6 Hz, ArH), 7.26–7.31 (4H, m, ArH), 8.05 (1H, d, J = 8.4 Hz, ArH), 9.40 (1H, br.s, NHCO). 13C NMR (CDCl3, 101 MHz): 24.3 (CH3), 40.6 (CH2), 58.9 (CH2), 114.9, 118.0 (ArCH), 119.3 (CH2), 119.8, 120.0, 123.1, 124.7, 125.6 (ArCH), 128.0 (ArC), 128.2, 128.2, 129.8 (ArCH), 130.7 (ArC), 134.0 (CH), 136.9, 139.3, 143.5, 156.1 (ArC), 168.1 (CO). Anal. Calcd. for C24H23ClN2O2. ½H2O C 69.31, H 5.82, N 6.74%. Found: C 68.9, H 5.75, N 6.50%. HRMS: Calcd. for C24H23ClN2O2 (M + H)+ 407.1521, found (M + H)+ 407.1503.
S-(+)-N-(2-(1-[2-(4-Chloro-phenoxy)-phenylamino]-but-2-enyl)-phenyl)-acetamide (32)
m.p. 139–141 °C (from hexane/DCM), LCMS (Chiracel AD-H column): tr = 14.8 min (80% MeOH in water), m/z M-H 405.4, HPLC (Chiracel AD-H column): tr = 11.50 min (90% acetonitrile in water), >99%, [α]D = +158.0, 1H NMR: As 191, 13C NMR: As 191, Anal. Calcd. for C24H23ClN2O2. ½H2O C 69.31, H 5.82, N 6.74%. Found: C 69.7, H 5.74, N 6.75%. HRMS: Calcd. for C24H23ClN2O2 (M + H)+ 407.1521, found (M + H)+ 407.1502. DCl3, 101 MHz): 20.2 (CH3), 50.1 (CH), 113.3, 116.6, 117.7, 118.6, 118.7, 119.3, 125.3, 126.7 (ArCH), 127.5, 127.7.


 ,
, 





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



