
Computational Design of High Energy RDX-Based Derivatives: Property Prediction, Intermolecular Interactions, and Decomposition Mechanisms


Abstract
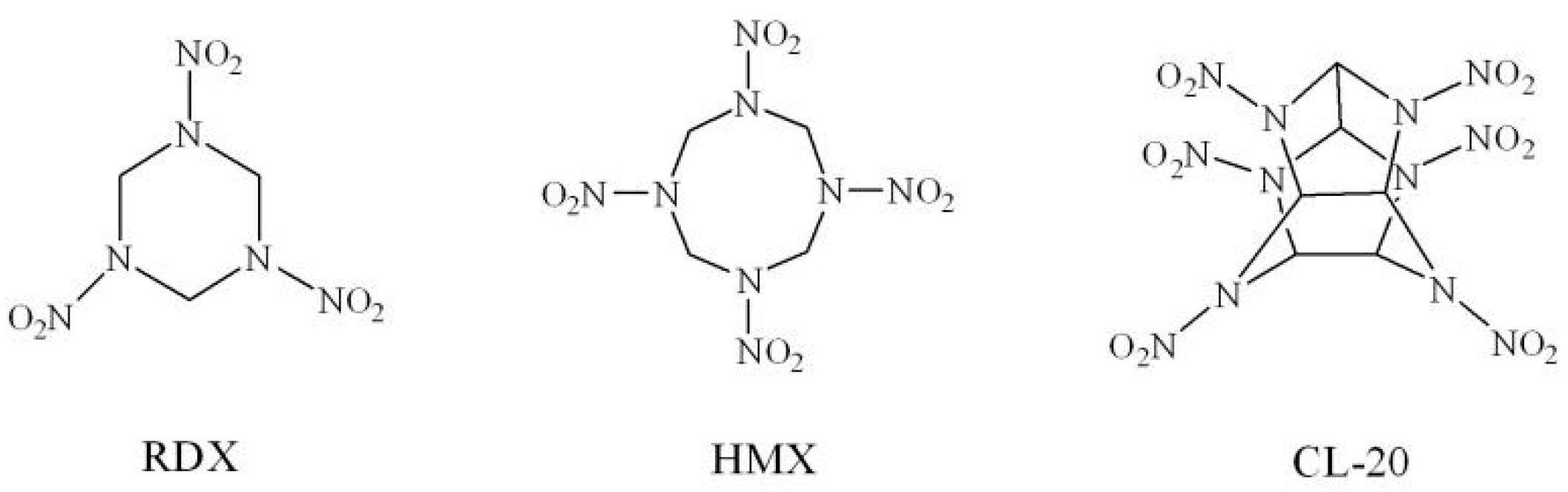
:1. Introduction
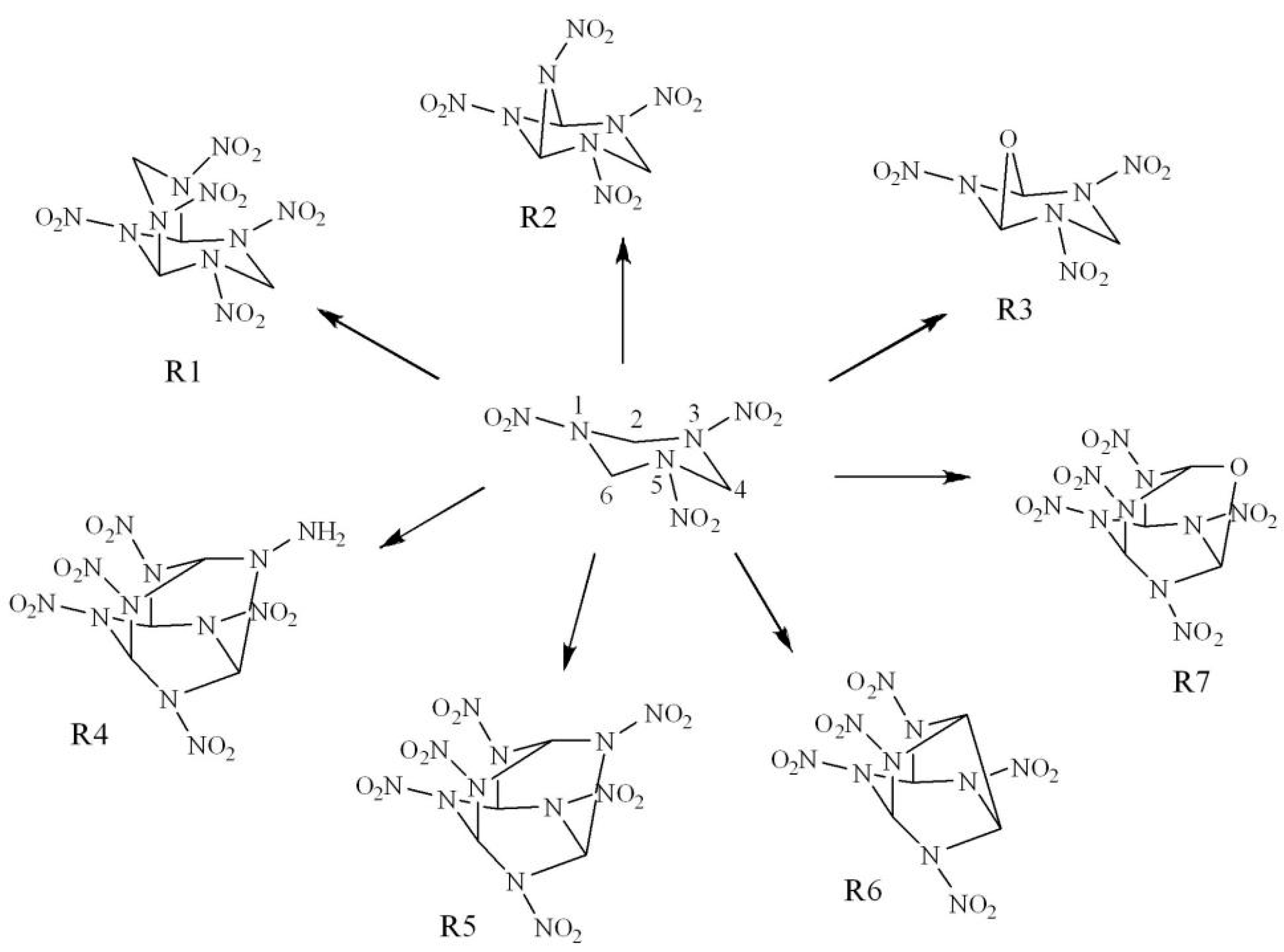
2. Results and Discussion
2.1. Property Prediction
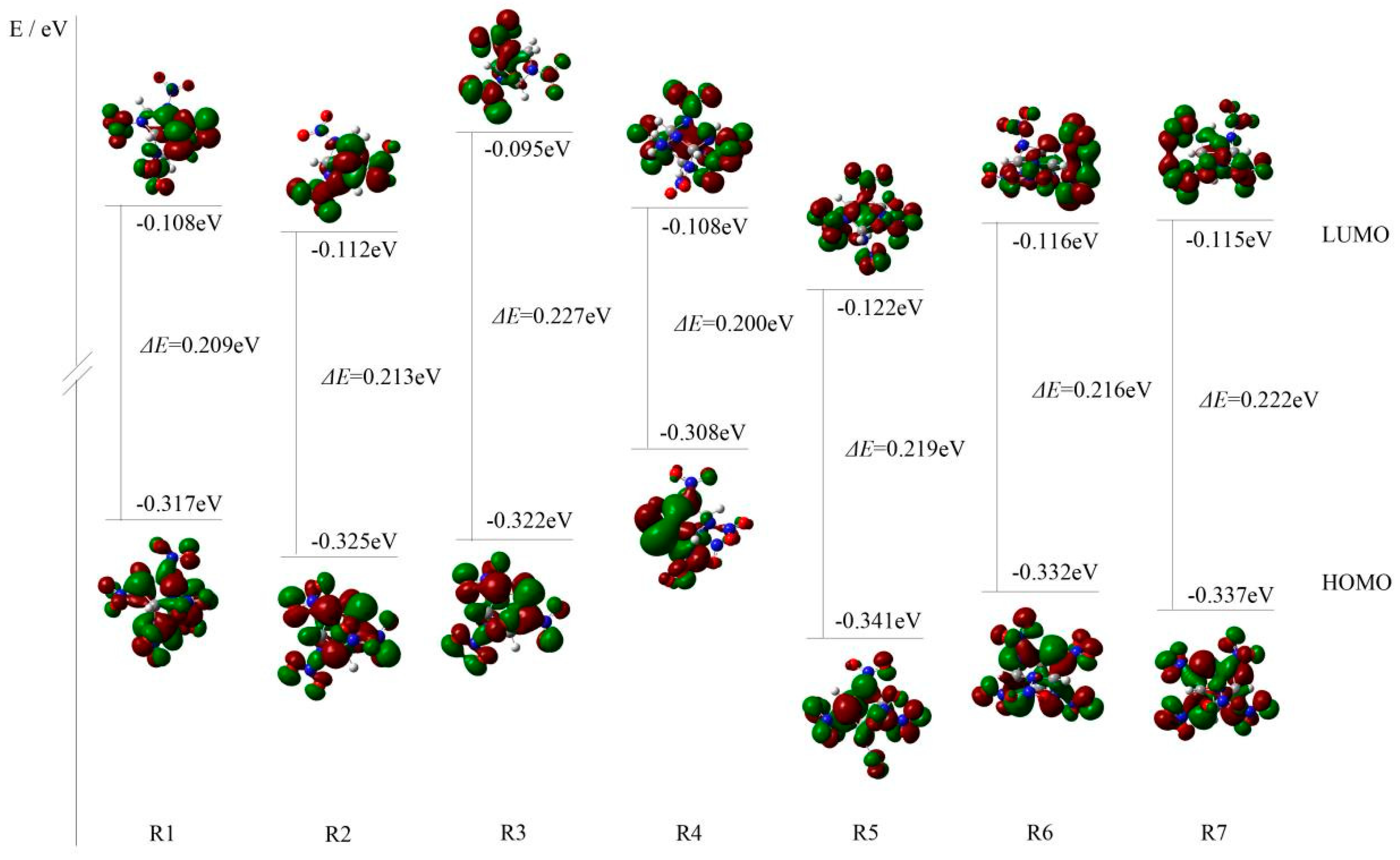
2.1.1. Molecular Geometry and Electronic Structures
2.1.2. HOFs
2.1.3. Detonation Properties
2.1.4. Impact Sensitivity
2.2. Molecular Packing
2.3. Intermolecular Interactions
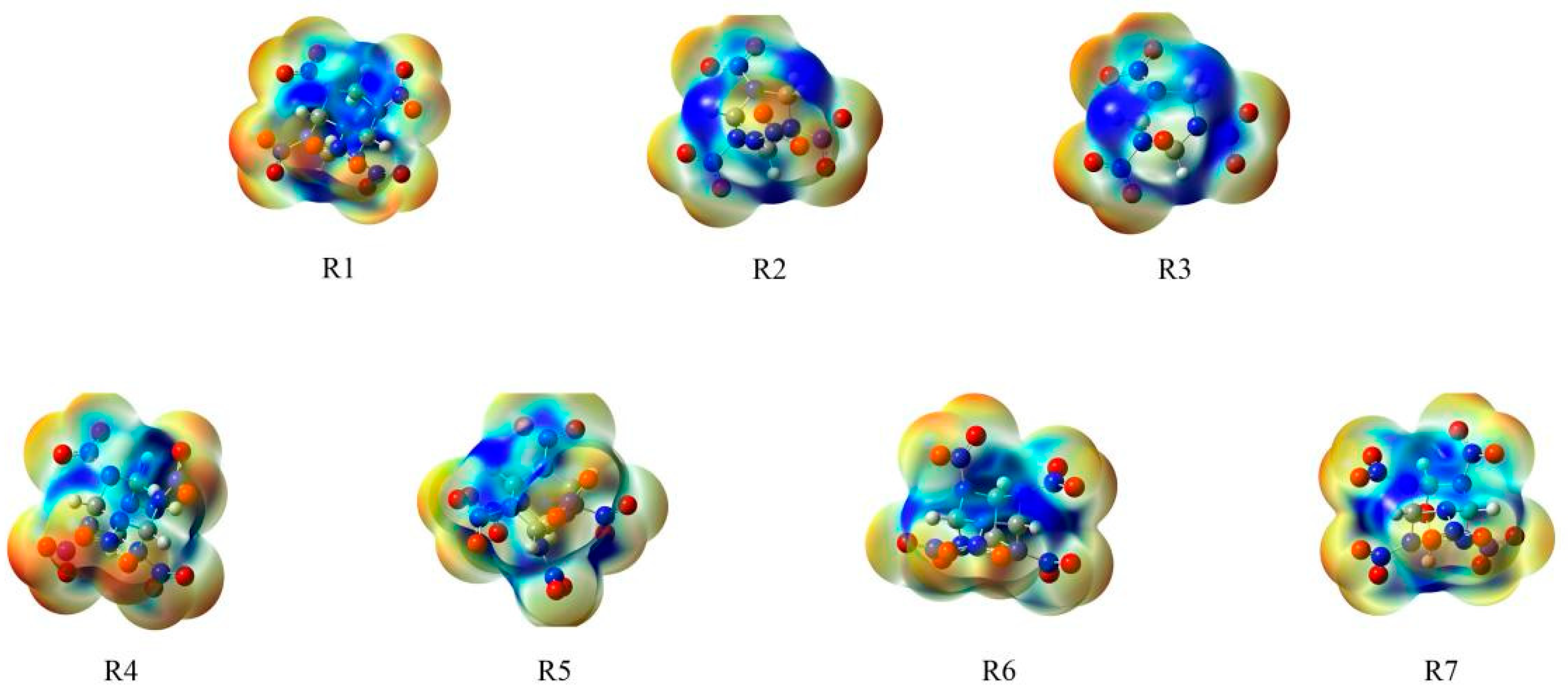
2.3.1. Geometrical Structures
2.3.2. Interaction Energy Analysis
2.3.3. AIM Topological Analysis
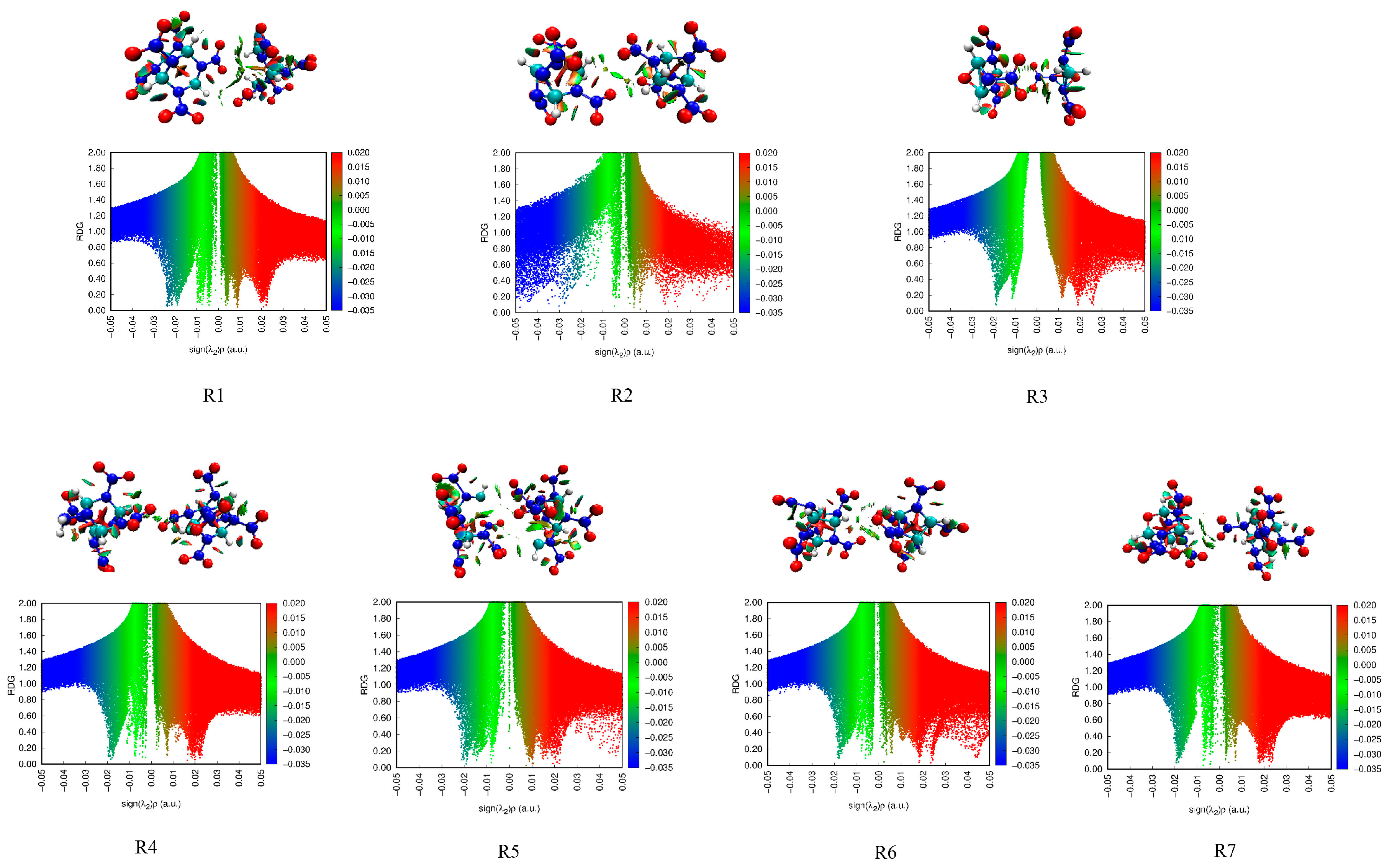
2.3.4. Reduced Density Gradients
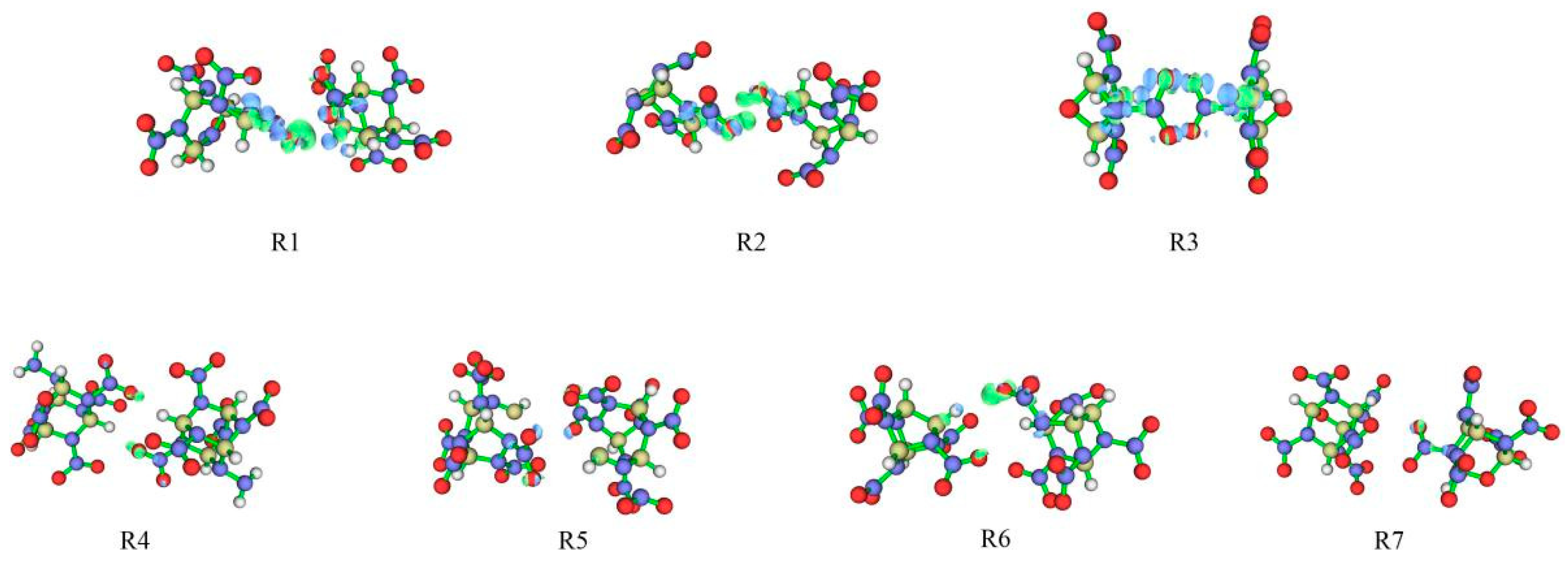
2.3.5. Electron Density Difference
2.4. Thermal Decomposition
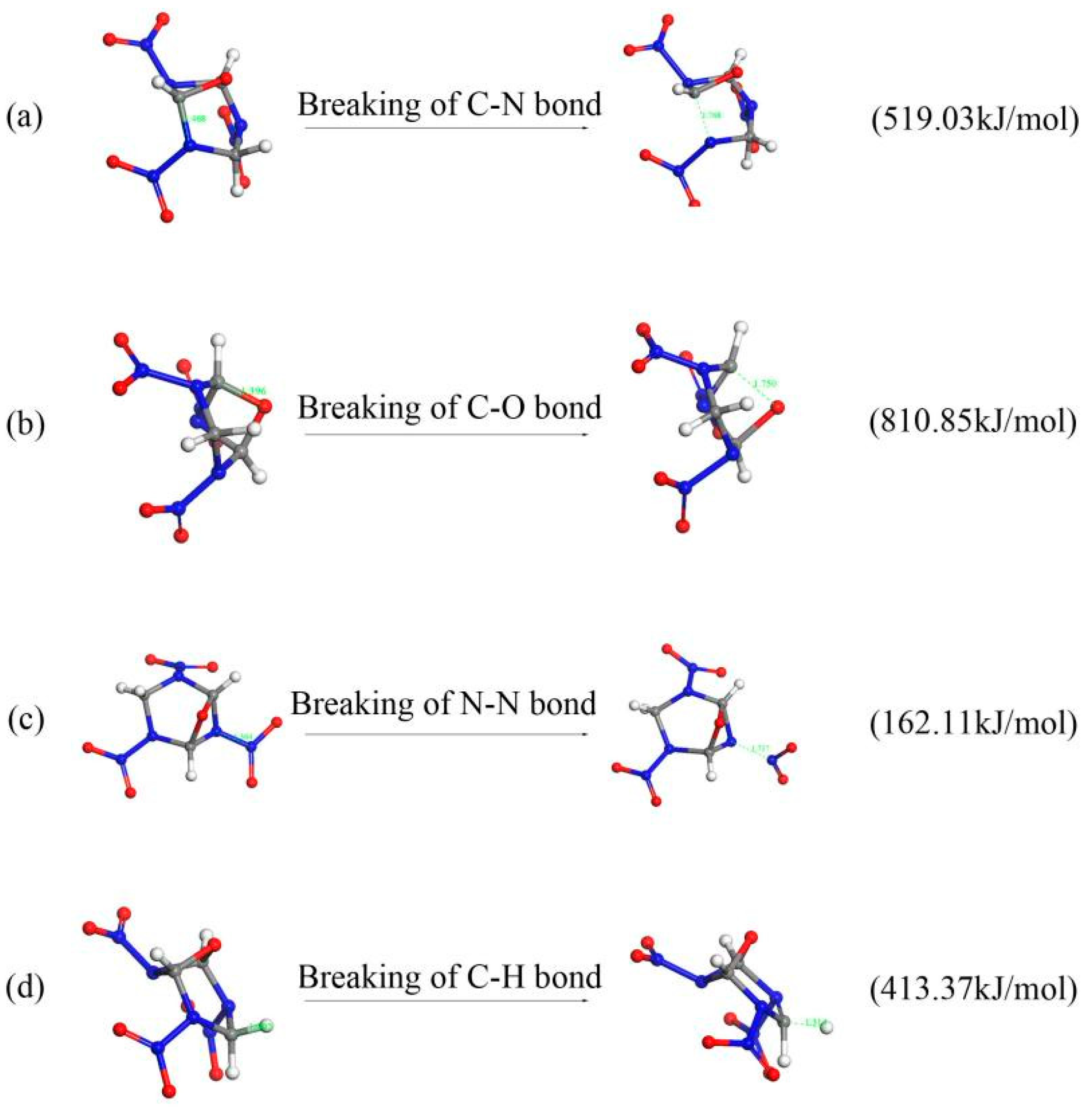
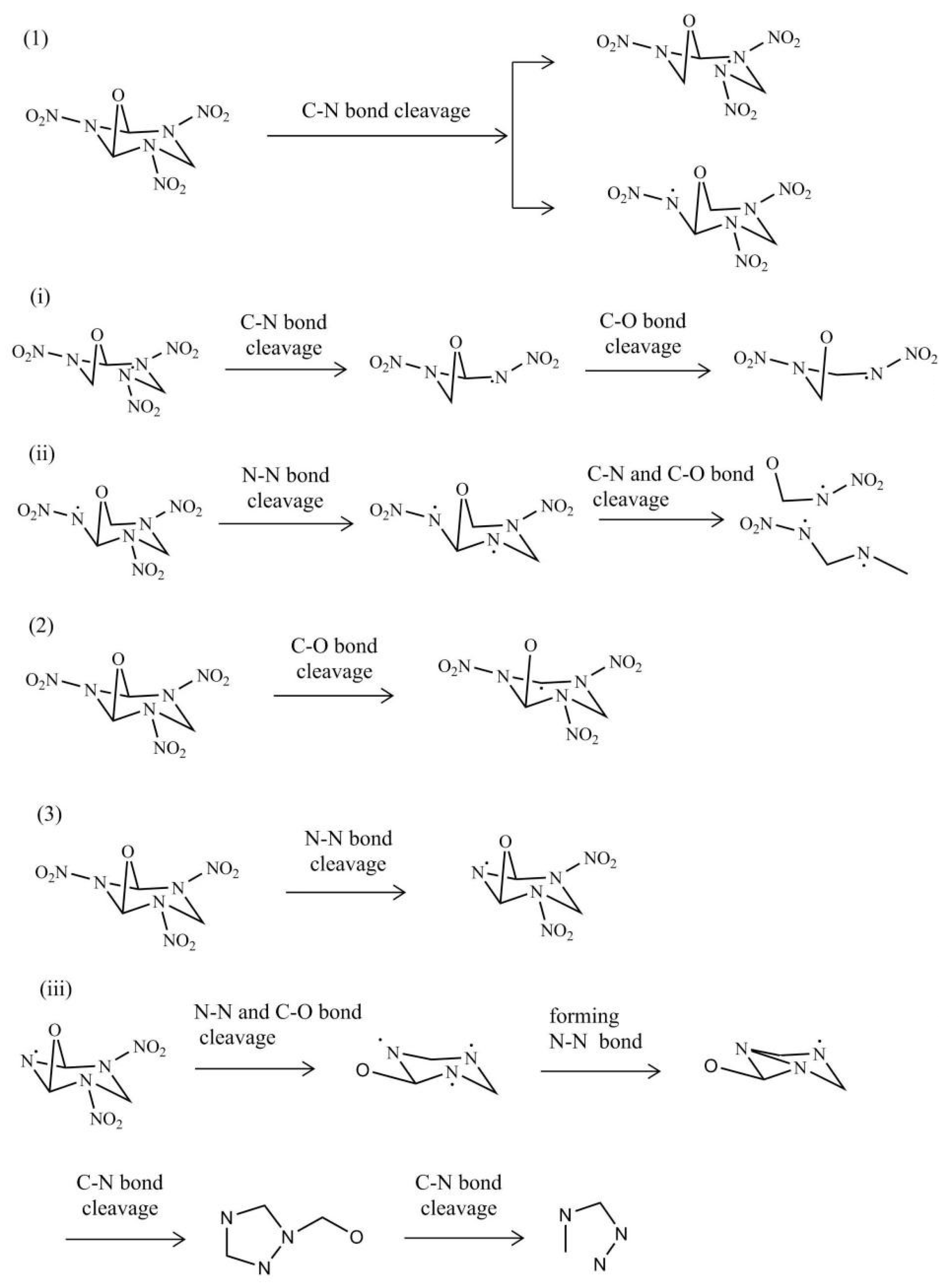
2.4.1. Initial Decomposition Mechanisms
2.4.2. Subsequent Decomposition
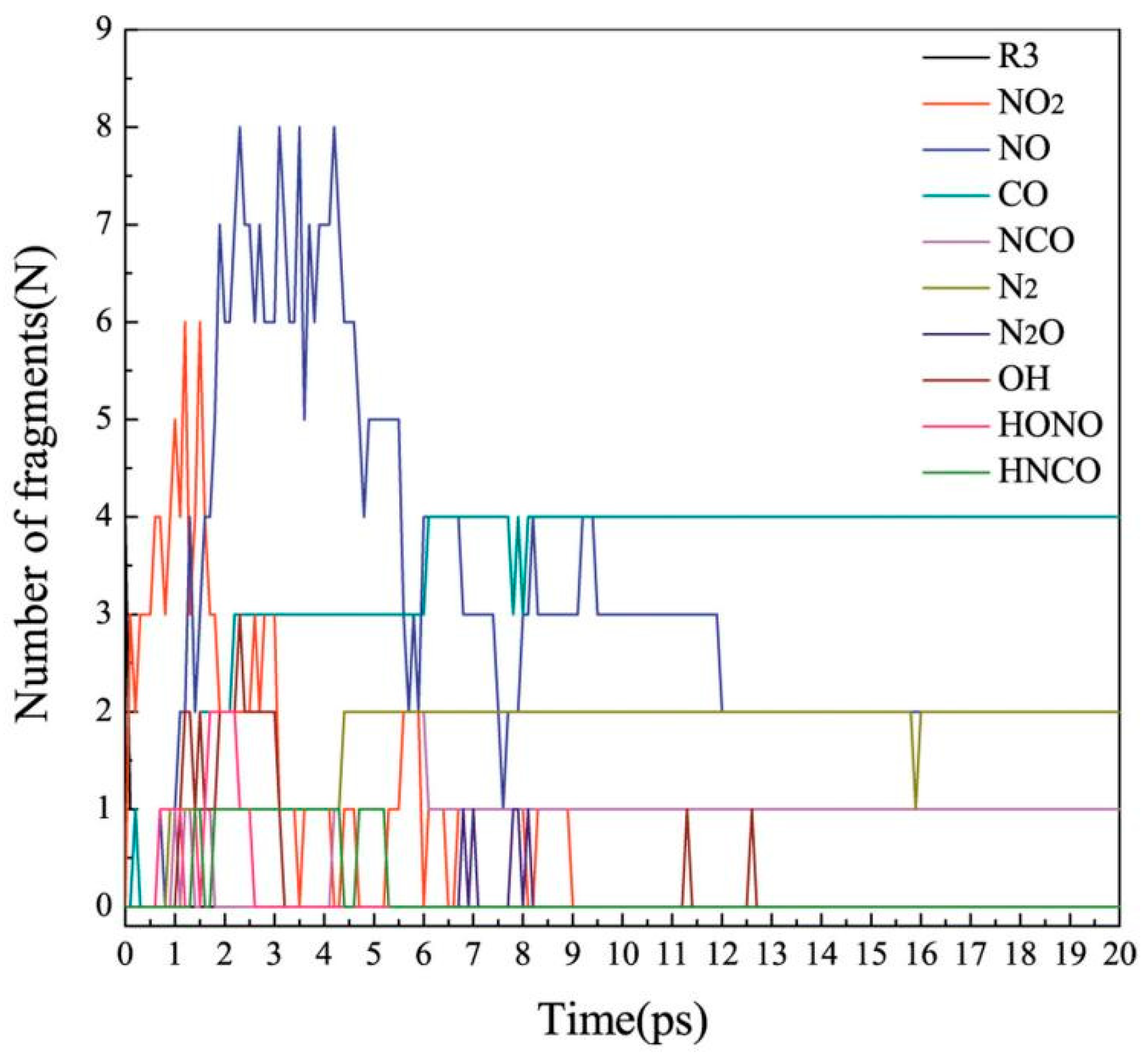
2.4.3. Decomposition Products
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Thottempudi, V.; Gao, H.X.; Shreeve, J.M. Trinitromethylsubstituted 5-Nitro- or 3-Azo-1,2,4-triazoles: Synthesis, Characterization, and Energetic Properties. J. Am. Chem. Soc. 2011, 133, 6464–6471. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tan, L.H.; Hang, Z.S.; Wang, J.Y.; Zhang, Z.W.; Zhu, W.H. A New Design Strategy on Cage Insensitive High Explosives: Symmetrically Replacing Carbon Atoms by Nitrogen Atoms Followed by the Introduction of N-oxides. RSC Adv. 2015, 5, 93607–93614. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, W.H.; Xiao, H.M. A New Design Strategy for High-energy Low-sensitivity Explosives: Combining Oxygen Balance Equal to Zero, A Combination of Nitro and Amino Groups, and Noxide in One Molecule of 1-amino-5-nitrotetrazole-3N-oxide. J. Mater. Chem. A 2014, 2, 13006–13015. [Google Scholar] [CrossRef]
- Sikder, A.K.; Sikder, N. A Review of Advanced High Performance, Insensitive and Thermally Stable Energetic Materials Emerging for Military and Space Applications. J. Hazard. Mater. 2004, 112, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Y.; Chi, W.J.; Li, Z.S.; Li, Q.S. Molecular Design of N-NO2 Substituted Cycloalkanes Derivatives Cm(N-NO2)m for Energetic Materials with High Detonation Performance and Low Impact Sensitivity. RSC Adv. 2015, 5, 38048–38055. [Google Scholar] [CrossRef]
- Wang, F.; Wang, G.X.; Du, H.C.; Zhang, J.Y.; Gong, X.D. Theoretical Studies on the Heats of Formation, Detonation Properties, and Pyrolysis Mechanisms of Energetic Cyclic Nitramines. J. Phys. Chem. A 2011, 115, 13858–13864. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Gong, X.D.; Ju, X.H.; Xiao, H.M. Substituent Effect on the Molecular Stability, Group Interaction, Detonation Performance, and Thermolysis Mechanism of Nitroamino-substituted Cyclopentanes and Cyclohexanes. Sci. China. Ser. B-Chem. 2008, 51, 1231–1245. [Google Scholar] [CrossRef]
- Qiu, L.; Xiao, H.M.; Gong, X.D.; Ju, X.H. Theoretical Studies on the Thermodynamic Properties and Detonation Performances of Bicyclic Nitramines: TNAD Isomers. Chin. J. Struc. Chem. 2006, 25, 1309–1320. [Google Scholar]
- Pan, Y.; Zhu, W.H.; Xiao, H.M. DFT Studies on Trinitromethylor Dinitromethyl-modified Derivatives of RDX and β-HMX. Comput. Theor. Chem. 2013, 1019, 116–124. [Google Scholar] [CrossRef]
- Du, M.R.; Wang, X.G.; Guo, Z.R. Theoretical Design of Bicyclo[2.2.1]heptane Derivatives for High-energy Density Compounds with Low Impact Sensitivity. Comput. Theor. Chem. 2016, 1095, 54–64. [Google Scholar] [CrossRef]
- Jin, X.H.; Hu, B.C.; Lu, W.; Gao, S.J.; Liu, Z.L.; Lv, C.X. Theoretical Study on a Novel High-energy Density Material 4,6,10,12-Tetranitro-5,11-bis(nitroimino)-2,8-dioxa-4,6,10,12-tetraaza-tricyclo-[7,3,0,03,7]dodecane. RSC Adv. 2014, 4, 6471–6477. [Google Scholar] [CrossRef]
- Qiu, L.; Xiao, H.M.; Gong, X.D.; Ju, X.H.; Zhu, W.H. Theoretical Studies on the Structures, Thermodynamic Properties, Detonation Properties, and Pyrolysis Mechanisms of Spiro Nitramines. J. Phys. Chem. A 2006, 110, 3797–3807. [Google Scholar] [CrossRef]
- Shen, C.; Wang, P.C.; Lu, M. Molecular Design and Property Prediction for a Series of Novel Dicyclic Cyclotrimethylene Trinitramines (RDX) Derivatized as High Energy Density Materials. J. Phys. Chem. A 2015, 119, 8250–8255. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, M.; Vávra, P.; Concha, M.C.; Murray, J.S.; Politer, P. A possible Crystal Volume Factor in the Impact Sensitivities of Some Energetic Compounds. J. Mol. Model. 2010, 16, 895–901. [Google Scholar] [CrossRef]
- Talawar, M.B.; Sivabalan, R.; Mukundan, T.; Muthurajan, H.; Sikder, A.K.; Gandhe, B.R.; Rao, A.S. Environmentally Compatible Next Generation Green Energetic Materials (GEMs). J. Hazard. Mater. 2009, 161, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Xiao, H.M.; Ju, X.H.; Gong, X.D.; Zhu, W.H. Computational Studies on Polynitrohexaazaadmantanes as Potential High Energy Density Materials. J. Phys. Chem. A 2006, 110, 5929–5933. [Google Scholar] [CrossRef] [PubMed]
- Ghule, V.D.; Jadhav, P.M.; Patil, R.S.; Radhakrishnan, S.; Soman, T. Quantum-Chemical Studies on Hexaazaisowurtzitanes. J. Phys. Chem. A 2010, 114, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhu, W.H. Theoretical Design on a Series of Novel Bicyclic and Cage Nitramines as High Energy Density Compounds. J. Phys. Chem. A 2017, 121, 9163–9171. [Google Scholar] [CrossRef]
- Thottempudi, V.; Shreeve, J.M. Synthesis and Promising Properties of a New Family of High-Density Energetic Salts of 5-Nitro-3-trinitromethyl-1H-1,2,4-triazole and 5,5′-Bis(trinitromethyl)-3,3′-azo-1H-1,2,4-triazole. J. Am. Chem. Soc. 2011, 133, 19982–19992. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Badgujar, D.M.; Talawar, M.B.; Asthana, S.N.; Mahulikar, P.P. Advances in Science and Technology of Modern Energetic Materials: An Overview. J. Hazard. Mater. 2008, 151, 289–305. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Schultz, P.A.; Desjarlais, M.P.; Mattsson, T.R.; Leung, K. Designing Meaningful Density Functional Theory Calculations in Materials Science-A Primer. Modell. Simul. Mater. Sci. Eng. 2005, 13, R1. [Google Scholar] [CrossRef]
- Xiang, D.; Zhu, W.H. Thermal Decomposition of Isolated and Crystal 4,10-Dinitro-2,6,8,12-tetraoxa-4,10-diazaisowurtzitane According to Ab Initio Molecular Dynamics Simulations. RSC Adv. 2017, 7, 8347–8356. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhu, W.H.; Xiao, H.M. Ab Initio Molecular Dynamics Study of Thermal Decomposition of 3,6-Di(azido)-1,2,4,5-tetrazine. Phys. Chem. Chem. Phys. 2014, 16, 21620–21628. [Google Scholar] [CrossRef]
- Rice, B.M.; Hare, J.J. A Quantum Mechanical Investigation of the Relation between Impact Sensitivity and the Charge Distribution in Energetic Molecules. J. Phys. Chem. A 2002, 106, 1770–1783. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Du, H.C.; Wang, F.; Gong, X.D.; Huang, Y.S. DFT Studies on a High Energy Density Cage Compound 4-Trinitroethyl-2,6,8,10,12-pentanitrohezaazaisowurtzitane. J. Phys. Chem. A 2011, 115, 6617–6621. [Google Scholar] [CrossRef]
- Li, J.S. Relationships for the Impact Sensitivities of Energetic C-Nitro Compounds Based on Bond Dissociation Energy. J. Phys. Chem. B 2010, 114, 2198–2202. [Google Scholar] [CrossRef]
- Mendoza-Cortes, J.L.; An, Q.; Goddard, W.A.; Ye, C.C.; Zybin, S. Prediction of the Crystal Packing of Di-tetrazine-tetraoxide (DTTO) Energetic Material. J. Comput. Chem. 2016, 37, 163–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.C.; Liu, Y.; Li, J.S.; Zhu, W.H. Prediction of Supramolecular Synthons and Crystal Packings of Supermolecular HMX/solvent Assemblies. RSC Adv. 2017, 7, 55482–55488. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar]
- Scheiner, S.; Kar, T. Red-versus Blue-Shifting Hydrogen Bonds: Are There Fundamental Distinctions. J. Phys. Chem. A 2002, 106, 1784–1789. [Google Scholar] [CrossRef]
- Pan, Y.; Li, J.S.; Cheng, B.B.; Zhu, W.H.; Xiao, H.M. Computational Studies on the Heats of Formation, Energetic Properties, and Thermal Stability of Energetic Nitrogen-rich Furazano-[3,4-b]pyrazine-based Derivatives. Comput. Theor. Chem. 2012, 992, 110–119. [Google Scholar] [CrossRef]
- Fan, X.W.; Ju, X.H. Theoretical Studies on Four-membered Ring Compounds with NF2, ONO2, N3, and NO2 Groups. J. Comput. Chem. 2008, 29, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Zhu, W.H.; Zhang, X.W.; Li, Y.F.; Xiao, H.M. Molecular Design of 1,2,4,5-Tetrazine-Based High-Energy Density Materials. J. Phys. Chem. A 2009, 113, 9404–9412. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Zhu, W.H.; Zhang, J.J.; Xiao, H.M. DFT Study on Energetic Tetrazolo-[1,5-b]-1,2,4,5-tetrazine and 1,2,4-Triazolo-[4,3-b]-1,2,4,5-tetrazine Derivatives. J. Hazard. Mater. 2010, 179, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhu, W.H.; Xiao, H.M. Design and Selection of Nitrogen-rich Bridged Di-1,3,5-triazine Derivatives with High Energy and Reduced Sensitivity. J. Mol. Model. 2012, 18, 3125–3138. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhu, W.H.; Xiao, H.M. Theoretical Studies on the Structures, Heats of Formation, Energetic Properties and Pyrolysis Mechanisms of Nitrogen-rich Difurazano[3,4-b:3′,4′-e]piperazine Derivatives and Their Analogues. Struct. Chem. 2013, 24, 1071–1087. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Some Perspectives on Estimating Detonation Properties of C, H, N, O Compounds. Cent. Eur. J. Energy Mater. 2011, 8, 209–220. [Google Scholar]
- Atkins, P.W. Physical Chemistry; Oxford University Press: Oxford, UK, 1982. [Google Scholar]
- Politzer, P.; Lane, P.; Murray, J.S. Computational Characterization of a Potential Energetic Compound: 1,3,5,7-Tetranitro-2,4,6,8-tetraazacubane. Cent. Eur. J. Energy Mater. 2011, 8, 39–52. [Google Scholar]
- Byrd, E.F.C.; Rice, B.M. Improved Prediction of Heats of Formation of Energetic Materials Using Quantum Mechanical Calculations. J. Phys. Chem. A 2006, 110, 1005–1013. [Google Scholar] [CrossRef]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative Analysis of Molecular Surfaces: Areas, Volumes, Electrostatic Potentials and Average Local Ionization Energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Jaidann, M.; Roy, S.; Abou-Rachid, H.; Lussier, L.S. A DFT Theoretical Study of Heats of Formation and Detonation Properties of Nitrogen-rich Explosives. J. Hazard. Mater. 2010, 176, 165–173. [Google Scholar] [CrossRef]
- Wang, F.; Du, H.C.; Zhang, J.Y.; Gong, X.D. Comparative Theoretical Studies of Energetic Azo s-Triazines. J. Phys. Chem. A 2011, 115, 11852–11860. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Martinez, J.; Murray, J.S.; Concha, M.C.; Toro-Labbé, A. An Electrostatic Interaction Correction for Improved Crystal Density Prediction. Mol. Phys. 2009, 107, 2095–2101. [Google Scholar] [CrossRef]
- Rice, B.M.; Byrd, E.F.C. Evaluation of Electrostatic Descriptors for Predicting Crystalline Density. J. Comput. Chem. 2013, 34, 2146–2151. [Google Scholar] [CrossRef] [PubMed]
- Kamlet, M.J.; Jacobs, S.J. Chemistry of detonation. I. A Simple Method for Calculation Detonation Properties of C–H–N–O Explosives. J. Chem. Phys. 1968, 48, 23–35. [Google Scholar] [CrossRef]
- Pospíšil, M.; Vávra, P.; Concha, M.C.; Murray, J.S.; Politzer, P. Sensitivity and the Available Free Space per Molecule in the Unit Cell. J. Mol. Model. 2011, 17, 2569–2574. [Google Scholar] [CrossRef]
- Keshavarz, M.H. Simple Relationship for Predicting Impact Sensitivity of Nitroaromatics, Nitramines, and Nitroaliphatics. Propellants Explos. Pyrotech. 2010, 35, 175–181. [Google Scholar] [CrossRef]
- Li, J.S. A Multivariate Relationship for the Impact Sensitivities of Energetic N-nitrocompounds Based on Bond Dissociation Energy. J. Hazard. Mater. 2010, 174, 728–733. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced errors. Mol. Phys. 2006, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles Simulation: Ideas, Illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, W.H.; Xiao, H.M. Structural Transformations and Absorption Properties of Crystalline 7-Amino-6-nitrobenzodifuroxan under High Pressures. J. Phys. Chem. C 2013, 117, 16830–16839. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Manaa, M.R.; Fried, L.E.; Melius, C.F.; Elstner, M.; Frauenheim, T. Decomposition of HMX at extreme conditions: A molecular dynamics simulation. J. Phys. Chem 2002, 106, 9024–9029. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ρ (g/cm3) | Q (cal/g) | D (km/s) | P (GPa) | OB 1 | h50 (cm) |
|---|---|---|---|---|---|---|
| R1 | 1.78 | 2152.91 | 9.63 | 40.86 | −4.52 | 33.16 |
| R2 | 1.88 | 2291.20 | 10.20 | 47.33 | 0.00 | 25.27 |
| R3 | 1.82 | 2089.75 | 9.65 | 41.67 | −6.78 | 40.73 |
| R4 | 1.94 | 2138.05 | 10.23 | 48.46 | −4.19 | 17.32 |
| R5 | 1.98 | 2019.73 | 10.19 | 48.67 | 7.77 | 13.87 |
| R6 | 1.94 | 2223.84 | 10.27 | 48.91 | 0.00 | 13.90 |
| R7 | 1.98 | 2006.87 | 10.14 | 48.13 | 4.35 | 19.68 |
| RDX | 1.77 (1.82 2) | 1559.99 (1501.00 3) | 8.78 (8.70 2) | 33.80 (34.00 2) | −21.62 | 26.00 |
| HMX | 1.84 (1.91 2) | 1532.78 (1498.00 3) | 9.01 (9.10 2) | 36.58 (39.30 2) | −21.62 | 30.00 |
| CL-20 | 1.94 (2.04 2) | 1671.87 (1738.20 3) | 9.44 (9.40 2) | 41.33 (42.00 2) | −10.96 | 11.94 |
| Assemble | Interaction | d | ρ(r) × 102 | ∇2ρ(r) × 102 | Gb(r) × 102 | Vb(r) × 102 | Hb(r) × 103 |
|---|---|---|---|---|---|---|---|
| R1-R1 | H(34)···O(17) | 2.70 | 0.5023 | 2.0030 | 0.4067 | −0.3126 | 0.9407 |
| H(30)···O(47) | 2.71 | 0.5177 | 2.1224 | 0.4342 | −0.3378 | 0.9640 | |
| H(29)···O(47) | 2.60 | 0.7653 | 2.7323 | 0.5946 | −0.5061 | 0.8850 | |
| R2-R2 | H(42)···O(9) | 2.44 | 0.9488 | 3.2314 | 0.7340 | −0.6601 | 0.7390 |
| H(18)···O(32) | 2.50 | 0.8422 | 2.9014 | 0.6479 | −0.5704 | 0.7749 | |
| R3-R3 | H(40)···O(15) | 2.50 | 0.8863 | 2.9218 | 0.6633 | −0.5962 | 0.6713 |
| H(20)···O(36) | 2.50 | 0.8864 | 2.9222 | 0.6634 | −0.5963 | 0.6713 | |
| R4-R4 | H(37)···O(23) | 2.78 | 0.4044 | 1.7658 | 0.3425 | −0.2436 | 0.9893 |
| H(5)···O(55) | 2.78 | 0.4043 | 1.7654 | 0.3424 | −0.2435 | 0.9892 | |
| R5-R5 | O(44)···O(22) | 3.25 | 0.3514 | 1.6855 | 0.3526 | −0.2839 | 0.6875 |
| O(52)···O(14) | 3.25 | 0.3513 | 1.6851 | 0.3526 | −0.2838 | 0.6872 | |
| R6-R6 | H(33)···O(21) | 2.44 | 0.7850 | 2.8778 | 0.6371 | −0.5547 | 0.8237 |
| H(28)···O(52) | 2.86 | 0.2766 | 1.3043 | 0.2419 | −0.1577 | 0.8421 | |
| R7-R7 | H(5)···O(47) | 2.86 | 0.3955 | 1.7819 | 0.3393 | −0.2332 | 1.0616 |
| Assembles | R1 | R2 | R3 | R4 | R5 | R6 | R7 |
|---|---|---|---|---|---|---|---|
| E(int.) (kcal/mol) | −8.19 | −7.02 | −9.46 | −4.94 | −6.18 | −6.16 | −5.42 |
| E(int.) (kJ/mol) | −34.27 | −29.37 | −39.58 | −20.67 | −25.86 | −25.77 | −22.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, L.; Zhu, W. Computational Design of High Energy RDX-Based Derivatives: Property Prediction, Intermolecular Interactions, and Decomposition Mechanisms. Molecules 2021, 26, 7199. https://doi.org/10.3390/molecules26237199
Tang L, Zhu W. Computational Design of High Energy RDX-Based Derivatives: Property Prediction, Intermolecular Interactions, and Decomposition Mechanisms. Molecules. 2021; 26(23):7199. https://doi.org/10.3390/molecules26237199
Chicago/Turabian StyleTang, Li, and Weihua Zhu. 2021. "Computational Design of High Energy RDX-Based Derivatives: Property Prediction, Intermolecular Interactions, and Decomposition Mechanisms" Molecules 26, no. 23: 7199. https://doi.org/10.3390/molecules26237199
APA StyleTang, L., & Zhu, W. (2021). Computational Design of High Energy RDX-Based Derivatives: Property Prediction, Intermolecular Interactions, and Decomposition Mechanisms. Molecules, 26(23), 7199. https://doi.org/10.3390/molecules26237199