
Exploiting the Chiral Ligands of Bis(imidazolinyl)- and Bis(oxazolinyl)thiophenes—Synthesis and Application in Cu-Catalyzed Friedel–Crafts Asymmetric Alkylation

 , ,
, ,  , ,
, ,  and
and
Abstract
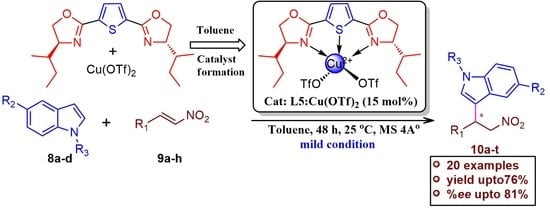
:
1. Introduction
2. Results and Discussion
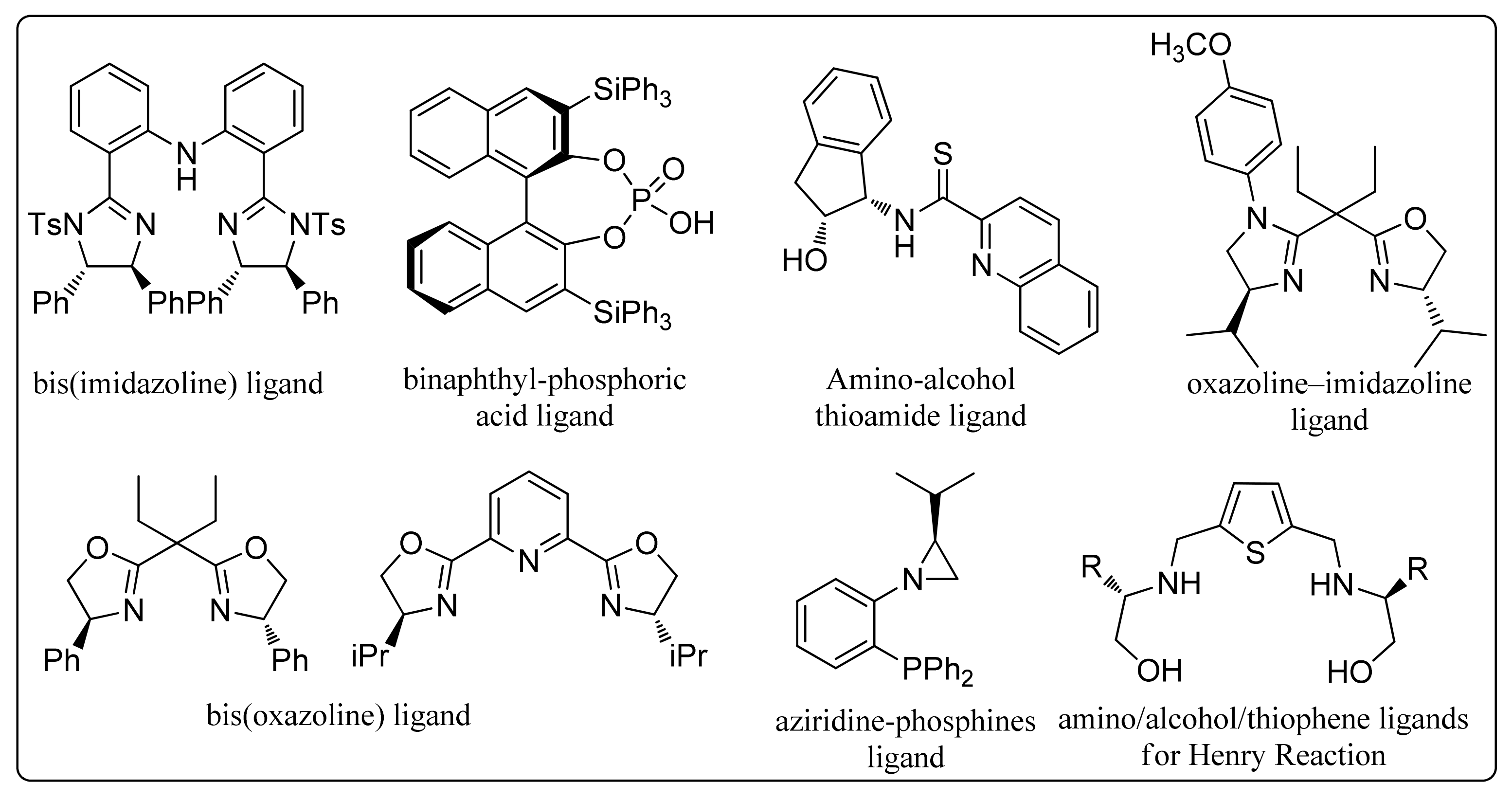
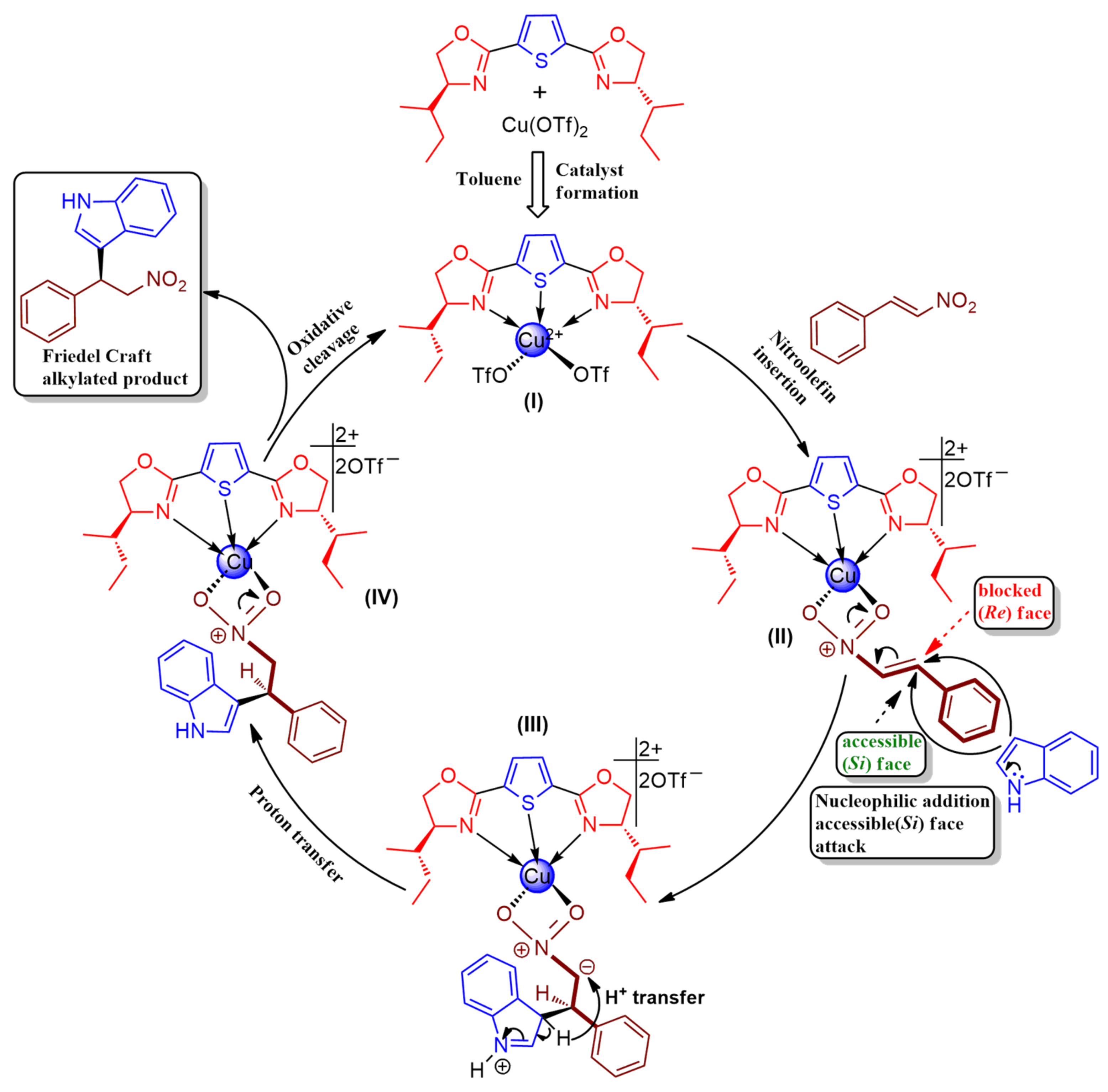
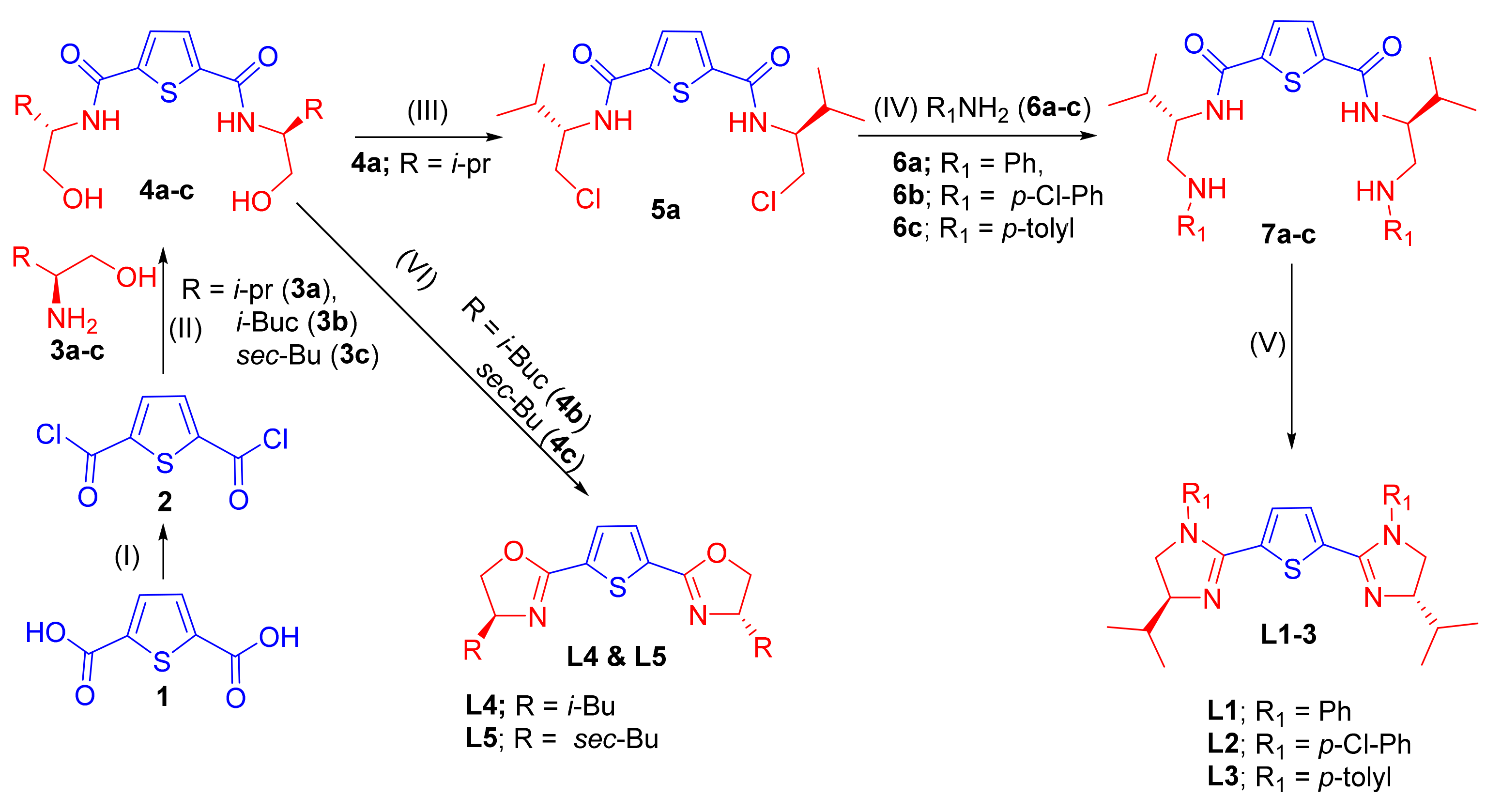
2.1. Synthesis of chiral 2,5-bis(imidazolinyl)thiophene (L1–L3) and 2,5-bis(oxazolinyl)thiophene (L4 and L5)
2.2. Application of Chiral Ligand (L1–L5)
2.2.1. Catalytic asymmetric Friedel–Crafts Alkylation of Indoles with Trans-β-nitrostyrene Derivatives; Optimization of Various Reaction Parameters
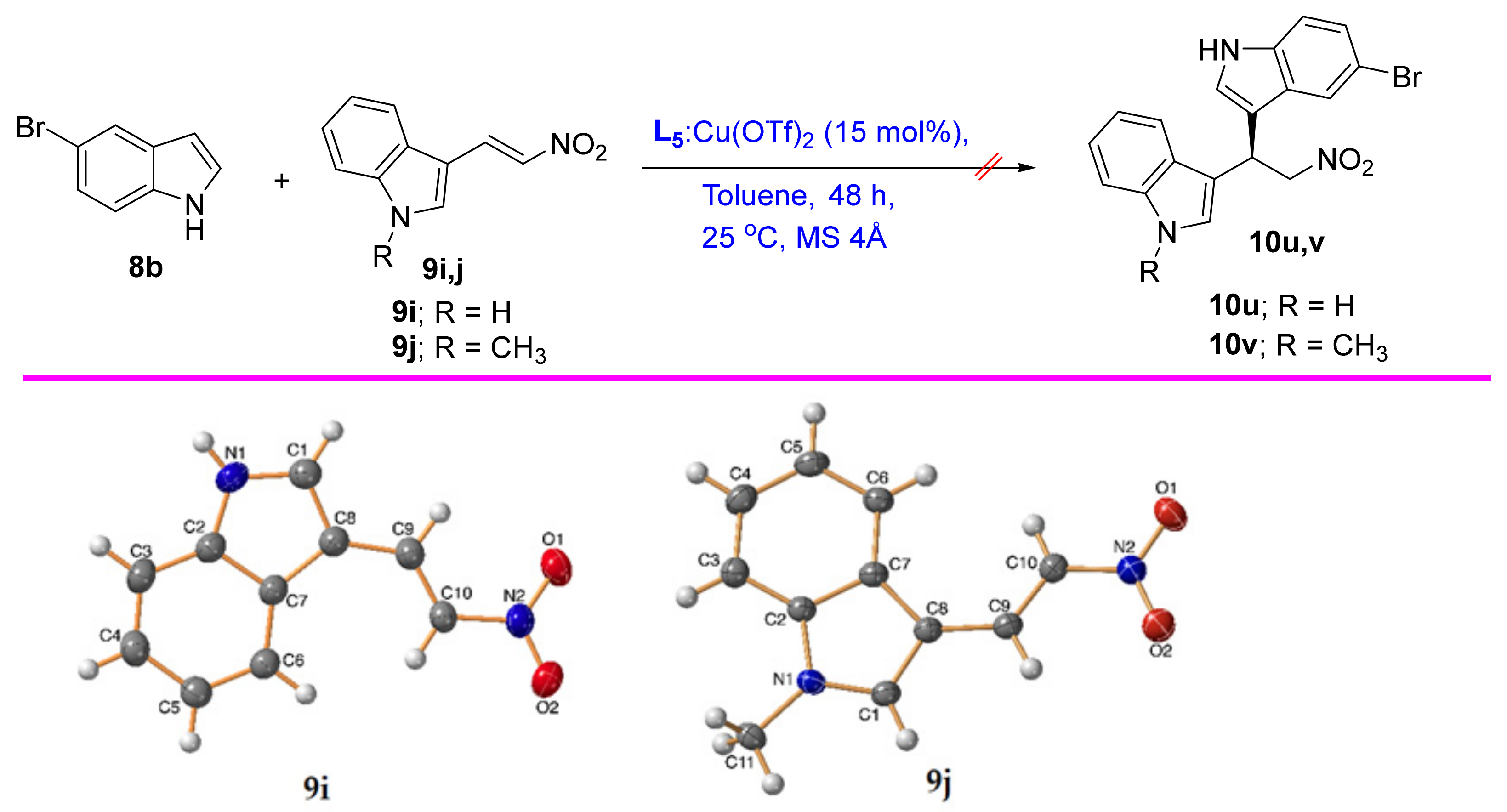
2.2.2. Substrate Scope
3. Materials and Methods
3.1. General
3.2. General Procedure (GP1) for the Preparation of Bis(hydroxyamides) 4a–c
3.2.1. N2,N5-Bis((S)-1-Hydroxy-3-methylbutan-2-yl)thiophene-2,5-dicarboxamide (4a)
3.2.2. N2,N5-Bis((S)-1-Hydroxy-4-methylpentan-2-yl)thiophene-2,5-dicarboxamide (4b)
3.2.3. N2,N5-Bis((2S,3R)-1-Hydroxy-3-methylpentan-2-yl)thiophene-2,5-dicarboxamide (4c)
3.3. General Procedure (GP2) for the Preparation of Thiophene-2,5-bis-imidazoline Chiral Ligands (L1–L3)
3.3.1. 2,5-Bis((S)-4-IsoPropyl-1-phenyl-4,5-dihydro-1H-imidazol-2-yl)thiophene (L1)
3.3.2. 2,5-Bis((S)-1-(4-Chlorophenyl)-4-isopropyl-4,5-dihydro-1H-imidazol-2-yl)thiophene (L2)
3.3.3. 2,5-Bis((S)-4-IsoPropyl-1-(p-tolyl)-4,5-dihydro-1H-imidazol-2-yl)thiophene (L3)
3.4. General Procedure (GP3) for the Synthesis of Thiophene-2,5-bis-oxazoline Chiral Ligands (L4 and L5)
3.4.1. 2,5-Bis((S)-4-isoButyl-4,5-dihydrooxazol-2-yl)thiophene (L4)
3.4.2. 2,5-Bis((S)-4-((S)-sec-Butyl)-4,5-dihydrooxazol-2-yl)thiophene (L5)
3.5. Synthesis of the β-nitrostyrene (9a–j)
3.6. Synthesis of Racemic Friedal–Crafts Alkylated Product Race-(10a–t)
3.7. General Procedure (GP4) for the Asymmetric Friedal–Crafts Alkylation of Indole to β-nitrostyrene (10a–t)
3.7.1. (S)-3-(1-(4-Fluorophenyl)-2-nitroethyl)-1H-indole (10a)
3.7.2. (S)-3-(1-(3-Bromophenyl)-2-nitroethyl)-1H-indole (10b)
3.7.3. (S)-3-(2-Nitro-1-(4-(trifluoromethyl)phenyl)ethyl)-1H-indole (10c)
3.7.4. (S)-3-(1-(4-Methoxyphenyl)-2-nitroethyl)-1H-indole (10d)
3.7.5. (R)-3-(2-Nitro-1-(2-nitrophenyl)ethyl)-1H-indole (10e)
3.7.6. (R)-3-(1-(2,4-Dichlorophenyl)-2-nitroethyl)-1H-indole (10f)
3.7.7. (S)-3-(2-Nitro-1-(thiophen-2-yl)ethyl)-1H-indole (10g)
3.7.8. (R)-3-(1-(2,6-Dichlorophenyl)-2-nitroethyl)-1H-indole (10h)
3.7.9. (S)-5-Bromo-3-(1-(4-fluorophenyl)-2-nitroethyl)-1H-indole (10i)
3.7.10. (S)-5-Bromo-3-(1-(3-bromophenyl)-2-nitroethyl)-1H-indole (10j)
3.7.11. (S)-5-Bromo-3-(2-nitro-1-(4-(trifluoromethyl)phenyl)ethyl)-1H-indole (10k)
3.7.12. (S)-5-Bromo-3-(1-(4-methoxyphenyl)-2-nitroethyl)-1H-indole (10l)
3.7.13. (R)-5-Bromo-3-(2-nitro-1-(2-nitrophenyl)ethyl)-1H-indole (10m)
3.7.14. (R)-5-Bromo-3-(1-(2,4-dichlorophenyl)-2-nitroethyl)-1H-indole (10n)
3.7.15. (S)-5-Bromo-3-(2-nitro-1-(thiophen-2-yl)ethyl)-1H-indole (10o)
3.7.16. (R)-5-Bromo-3-(1-(2,6-dichlorophenyl)-2-nitroethyl)-1H-indole (10p)
3.7.17. (S)-5-Fluoro-3-(2-nitro-1-(thiophen-2-yl)ethyl)-1H-indole(10q)
3.7.18. (R)-3-(1-(2,6-Dichlorophenyl)-2-nitroethyl)-1H-indole (10r)
3.7.19. (S)-1-Ethyl-3-(1-(4-fluorophenyl)-2-nitroethyl)-1H-indole (10s)
3.7.20. (S)-1-Ethyl-3-(1-(4-methoxyphenyl)-2-nitroethyl)-1H-indole (10t)
4. Large-Scale Synthesis of (S)-3-(1-(4-Fluorophenyl)-2-nitroethyl)-1H-indole (10a)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, F.; Pu, X.; Qi, X.; Lynch, V.; Radha, A.; Ready, J.M. Chiral allene-containing phosphines in asymmetric catalysis. J. Am. Chem. Soc. 2011, 133, 18066–18069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeuwen, P.W.v.; Kamer, P.C.; Claver, C.; Pamies, O.; Dieguez, M. Phosphite-containing ligands for asymmetric catalysis. Chem. Rev. 2011, 111, 2077–2118. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pérez, H.; Etayo, P.; Panossian, A.; Vidal-Ferran, A. Phosphine− phosphinite and phosphine− phosphite ligands: Preparation and applications in asymmetric catalysis. Chem. Rev. 2011, 111, 2119–2176. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Buzas, A.K.; Besnard, C.l.; Kündig, E.P. Chiral n-heterocyclic carbene gold complexes: Synthesis, properties, and application in asymmetric catalysis. Organometallics 2012, 31, 8348–8354. [Google Scholar] [CrossRef]
- Yoon, M.; Srirambalaji, R.; Kim, K. Homochiral metal–organic frameworks for asymmetric heterogeneous catalysis. Chem. Rev. 2012, 112, 1196–1231. [Google Scholar] [CrossRef]
- Chen, X.; Lu, Z. Recent advances in chiral imino-containing ligands for metal-catalyzed asymmetric transformations. Org. Biomol. Chem. 2017, 15, 2280–2306. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective iron-catalyzed transformations. Coord. Chem. Rev. 2019, 386, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Barakat, A.; El-Faham, A.; Haukka, M.; Al-Majid, A.M.; Soliman, S.M. S-triazine pincer ligands: Synthesis of their metal complexes, coordination behavior, and applications. Appl. Organomet. Chem. 2021, 35, e6317. [Google Scholar] [CrossRef]
- Kagan, H.B.; Gopalaiah, K. Early history of asymmetric synthesis: Who are the scientists who set up the basic principles and the first experiments? New J. Chem. 2011, 35, 1933–1937. [Google Scholar] [CrossRef]
- Oliveira, V.d.G.; Cardoso, M.F.d.C.; Forezi, L.d.S.M. Organocatalysis: A brief overview on its evolution and applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 38, 9267–9331. [Google Scholar] [CrossRef]
- Itoh, T.; Hanefeld, U. Enzyme catalysis in organic synthesis. Green Chem. 2017, 19, 331–332. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D.; Bode, M.L. The hitchhiker’s guide to biocatalysis: Recent advances in the use of enzymes in organic synthesis. Chem. Sci. 2020, 11, 2587–2605. [Google Scholar] [CrossRef] [Green Version]
- Pàmies, O.; Bäckvall, J.-E. Combination of enzymes and metal catalysts. A powerful approach in asymmetric catalysis. Chem. Rev. 2003, 103, 3247–3262. [Google Scholar] [CrossRef]
- Choi, J.; Fu, G.C. Catalytic asymmetric synthesis of secondary nitriles via stereoconvergent negishi arylations and alkenylations of racemic α-bromonitriles. J. Am. Chem. Soc. 2012, 134, 9102–9105. [Google Scholar] [CrossRef] [Green Version]
- Kalek, M.; Fu, G.C. Phosphine-catalyzed doubly stereoconvergent γ-additions of racemic heterocycles to racemic allenoates: The catalytic enantioselective synthesis of protected α, α-disubstituted α-amino acid derivatives. J. Am. Chem. Soc. 2015, 137, 9438–9442. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Lackey, H.H.; Ondrusek, B.A.; McQuade, D.T. Stereoconvergent synthesis of chiral allylboronates from an e/z mixture of allylic aryl ethers using a 6-NHC−Cu(I) catalyst. J. Am. Chem. Soc. 2011, 133, 2410–2413. [Google Scholar] [CrossRef]
- Li, L.; Chen, Z.; Zhang, X.; Jia, Y. Divergent strategy in natural product total synthesis. Chem. Rev. 2018, 118, 3752–3832. [Google Scholar] [CrossRef]
- Shimokawa, J. Divergent strategy in natural product total synthesis. Tetrahedron Lett. 2014, 55, 6156–6162. [Google Scholar] [CrossRef] [Green Version]
- Krautwald, S.; Carreira, E.M. Stereodivergence in asymmetric catalysis. J. Am. Chem. Soc. 2017, 139, 5627–5639. [Google Scholar] [CrossRef]
- Pellissier, H. Enantioselective vanadium-catalyzed transformations. An update. Coord. Chem. Rev. 2020, 418, 213395. [Google Scholar] [CrossRef]
- Roberts, R.M.; Khalaf, A.A. Friedel–Crafts Alkylation Chemistry: A Century of Discovery; Marcel Dekker Incorporated: New York, NY, USA, 1984; Volume 10. [Google Scholar]
- Olah, G.A. Friedel–Crafts and related reactions. In Across Conventional Lines: Selected Papers of George a Olah Volume 1; World Scientific: Singapore, 2003; pp. 109–118. [Google Scholar]
- Bandini, M.; Melloni, A.; Umani-Ronchi, A. New catalytic approaches in the stereoselective Friedel–Crafts alkylation reaction. Angew. Chem. Int. Ed. 2004, 43, 550–556. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A.; Umani-Ronchi, A. An update on catalytic enantioselective alkylations of indoles. Mini Rev. Org. Chem. 2007, 4, 115–124. [Google Scholar] [CrossRef]
- Bi, X.; Zhang, Q.; Gu, Z. Transition-metal-catalyzed carbon-carbon bond activation in asymmetric synthesis. Chin. J. Chem. 2021, 39, 1397–1412. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Al Majid, A.M.; Al-Othman, Z.A. Highly enantioselective Friedel–Crafts alkylation of indoles with α, β-unsaturated ketones with simple Cu(II)–oxazoline–imidazoline catalysts. Tetrahedron 2013, 69, 5185–5192. [Google Scholar] [CrossRef]
- Liu, L.; Ma, H.; Xiao, Y.; Du, F.; Qin, Z.; Li, N.; Fu, B. Highly enantioselective Friedel–Crafts alkylation of indoles and pyrrole with β, γ-unsaturated α-ketoesters catalyzed by heteroarylidene-tethered bis(oxazoline) copper complexes. Chem. Commun. 2012, 48, 9281–9283. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Kardak, B.G.; García, J.M.; Linden, A. Highly enantioselective friedel−crafts alkylations of pyrroles and indoles with α ‘-hydroxy enones under Cu(II)-simple bis(oxazoline) catalysis. J. Am. Chem. Soc. 2005, 127, 4154–4155. [Google Scholar] [CrossRef]
- Bedekar, A.V.; Andersson, P.G. A new class of bis-oxazoline ligands for the cu-catalysed asymmetric cyclopropanation of olefins. Tetrahedron Lett. 1996, 37, 4073–4076. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, X.; Shang, D.; Liu, X.; Feng, X. N, N′-dioxide–Scandium(III) complex catalyzed highly enantioselective Friedel–Crafts alkylation of indole to alkylidene malonates. Tetrahedron 2010, 66, 1447–1457. [Google Scholar] [CrossRef]
- Chen, H.; Du, F.; Liu, L.; Li, J.; Zhao, Q.; Fu, B. Malonate-type bis(oxazoline) ligands with sp2 hybridized bridge carbon: Synthesis and application in Friedel–Crafts alkylation and allylic alkylation. Tetrahedron 2011, 67, 9602–9608. [Google Scholar] [CrossRef]
- Zhou, J.; Ye, M.-C.; Huang, Z.-Z.; Tang, Y. Controllable enantioselective friedel−crafts reaction1 between indoles and alkylidene malonates catalyzed by pseudo-C3-symmetric trisoxazoline copper(II) complexes. J. Org. Chem. 2004, 69, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Fu, G.C. Nickel-catalyzed asymmetric negishi cross-couplings of secondary allylic chlorides with alkylzincs. J. Am. Chem. Soc. 2008, 130, 2756–2757. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Scheidt, K.A.; Fandrick, K.R.; Lam, H.W.; Wu, J. Enantioselective indole Friedel−Crafts alkylations catalyzed by bis (oxazolinyl) Pyridine−Scandium(III)triflate complexes. J. Am. Chem. Soc. 2003, 125, 10780–10781. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, M.; Li, Z.; Bian, Q.; Mao, J.; Li, S.; Liu, S.; Wang, M.; Zhong, J.; Guo, H. Asymmetric henry reaction catalyzed by a zn–amino alcohol system. Tetrahedron Asymmetry 2011, 22, 1156–1160. [Google Scholar] [CrossRef]
- Zhu, S.-F.; Xu, B.; Wang, G.-P.; Zhou, Q.-L. Well-defined binuclear chiral spiro copper catalysts for enantioselective n–h insertion. J. Am. Chem. Soc. 2012, 134, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Fu, G.C. Nickel/bis(oxazoline)-catalyzed asymmetric kumada reactions of alkyl electrophiles: Cross-couplings of racemic α-bromoketones. J. Am. Chem. Soc. 2010, 132, 1264–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, P.; Bera, P.K.; Noor-ul, H.K.; Kureshy, R.I.; Abdi, S.H.; Bajaj, H.C. Asymmetric Friedel–Crafts addition of indoles to n-sulfonyl aldimines catalyzed by Cu(II) chiral amino alcohol based schiff base complexes. Catal. Sci. Technol. 2014, 4, 563–568. [Google Scholar] [CrossRef]
- Ibáñez, I.; Kaneko, M.; Kamei, Y.; Tsutsumi, R.; Yamanaka, M.; Akiyama, T. Enantioselective Friedel–Crafts alkylation reaction of indoles with α-trifluoromethylated β-nitrostyrenes catalyzed by chiral binol metal phosphate. ACS Catal. 2019, 9, 6903–6909. [Google Scholar] [CrossRef]
- Islam, M.S.; Al Majid, A.M.; Al-Othman, Z.A.; Barakat, A. Highly enantioselective Friedel–Crafts alkylation of indole with electron deficient trans-β-nitroalkenes using Zn(II)–oxazoline–imidazoline catalysts. Tetrahedron Asymmetry 2014, 25, 245–251. [Google Scholar] [CrossRef]
- Singh, P.K.; Bisai, A.; Singh, V.K. Enantioselective Friedel–Crafts alkylation of indoles with nitroalkenes catalyzed by a bis(oxazoline)–cu (ii) complex. Tetrahedron Lett. 2007, 48, 1127–1129. [Google Scholar] [CrossRef]
- Li, W. Chiral bis(oxazolinyl) thiophenes for enantioselective Cu(II)-catalyzed Friedel–Crafts alkylation of indole derivatives with nitroalkenes. Catal. Lett. 2014, 144, 943–948. [Google Scholar] [CrossRef]
- Tanaka, K.; Sakuragi, K.; Ozaki, H.; Takada, Y. Highly enantioselective Friedel–Crafts alkylation of n, n-dialkylanilines with trans-β-nitrostyrene catalyzed by a homochiral metal–organic framework. Chem. Commun. 2018, 54, 6328–6331. [Google Scholar] [CrossRef]
- Li, Z.; He, M.; Xu, D.; Liu, Z. Graphene materials-based energy acceptor systems and sensors. J. Photochem. Photobiol. C. Photochem. Rev. 2014, 18, 1–17. [Google Scholar] [CrossRef]
- Gao, J.-R.; Wu, H.; Xiang, B.; Yu, W.-B.; Han, L.; Jia, Y.-X. Highly enantioselective construction of trifluoromethylated all-carbon quaternary stereocenters via nickel-catalyzed Friedel–Crafts alkylation reaction. J. Am. Chem. Soc. 2013, 135, 2983–2986. [Google Scholar] [CrossRef]
- Chen, J.-B.; Jia, Y.-X. Recent progress in transition-metal-catalyzed enantioselective indole functionalizations. Org. Biomol. Chem. 2017, 15, 3550–3567. [Google Scholar] [CrossRef]
- Ono, N. The Nitro Group in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2003; Volume 9. [Google Scholar]
- Aitken, L.S.; Arezki, N.R.; Dell’Isola, A.; Cobb, A.J. Asymmetric organocatalysis and the nitro group functionality. Synthesis 2013, 45, 2627–2648. [Google Scholar]
- Robinson, B. Alkaloids of the calabar bean. In The Alkaloids: Chemistry and Physiology; Elsevier: Amsterdam, The Netherlands, 1971; Volume 13, pp. 213–226. [Google Scholar]
- Takano, S.; Ogasawara, K. Alkaloids of the calabar bean. In The Alkaloids: Chemistry and Pharmacology; Elsevier: Amsterdam, The Netherlands, 1990; Volume 36, pp. 225–251. [Google Scholar]
- Greig, N.H.; Pei, X.F.; Soncrant, T.T.; Ingram, D.K.; Brossi, A. Phenserine and ring c hetero-analogues: Drug candidates for the treatment of alzheimer’s disease. Med. Res. Rev. 1995, 15, 3–31. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric michael additions to nitroalkenes. Eur. J. Org. Chem. 2002, 2002, 1877–1894. [Google Scholar] [CrossRef]
- Calderari, G.; Seebach, D. Asymmetrische michael-additionen. Stereoselektive alkylierung chiraler, nicht racemischer enolate durch nitroolefine. Herstellung enantiomerenreiner γ-aminobuttersäure-und bernsteinsäure-derivate. Helv. Chim. Acta 1985, 68, 1592–1604. [Google Scholar] [CrossRef]
- Hayashi, T.; Senda, T.; Ogasawara, M. Rhodium-catalyzed asymmetric conjugate addition of organoboronic acids to nitroalkenes. J. Am. Chem. Soc. 2000, 122, 10716–10717. [Google Scholar] [CrossRef]
- Choi, H.; Hua, Z.; Ojima, I. Highly enantioselective copper-catalyzed conjugate addition of diethylzinc to nitroalkenes. Org. Lett. 2004, 6, 2689–2691. [Google Scholar] [CrossRef]
- Duursma, A.; Minnaard, A.J.; Feringa, B.L. Highly enantioselective conjugate addition of dialkylzinc reagents to acyclic nitroalkenes: A catalytic route to β2-amino acids, aldehydes, and alcohols. J. Am. Chem. Soc. 2003, 125, 3700–3701. [Google Scholar] [CrossRef]
- Czekelius, C.; Carreira, E.M. Catalytic enantioselective conjugate reduction of β, β-disubstituted nitroalkenes. Angew. Chem. Int. Ed. 2003, 115, 4941–4943. [Google Scholar] [CrossRef]
- Watanabe, M.; Ikagawa, A.; Wang, H.; Murata, K.; Ikariya, T. Catalytic enantioselective michael addition of 1, 3-dicarbonyl compounds to nitroalkenes catalyzed by well-defined chiral ru amido complexes. J. Am. Chem. Soc. 2004, 126, 11148–11149. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Tang, L.; Deng, L. Highly enantioselective conjugate addition of malonate and β-ketoester to nitroalkenes: Asymmetric c−c bond formation with new bifunctional organic catalysts based on cinchona alkaloids. J. Am. Chem. Soc. 2004, 126, 9906–9907. [Google Scholar] [CrossRef]
- Mellah, M.; Voituriez, A.; Schulz, E. Chiral sulfur ligands for asymmetric catalysis. Chem. Rev. 2007, 107, 5133–5209. [Google Scholar] [CrossRef]
- Alammari, A.S.; Al-Majid, A.M.; Barakat, A.; Alshahrani, S.; Ali, M.; Islam, M.S. Asymmetric henry reaction of nitromethane with substituted aldehydes catalyzed by novel in situ generated chiral bis(β-amino alcohol-cu(oac)2·h2o complex. Catalysts 2021, 11, 1208. [Google Scholar] [CrossRef]
- Mao, J.; Nie, X.; Wang, M.; Wang, Q.; Zheng, B.; Bian, Q.; Zhong, J. Catalytic asymmetric nitroaldol (henry) reactions with copper(ii)/cyclopropane-based bisoxazoline complexes. Tetrahedron Asymmetry 2012, 23, 965–971. [Google Scholar] [CrossRef]
- Blay, G.; Climent, E.; Fernandez, I.; Hernández-Olmos, V.; Pedro, J.R. Enantioselective henry reaction catalyzed with copper(II)–iminopyridine complexes. Tetrahedron Asymmetry 2007, 18, 1603–1612. [Google Scholar] [CrossRef]
- Hao, X.-Q.; Xu, Y.-X.; Yang, M.-J.; Wang, L.; Niu, J.-L.; Gong, J.-F.; Song, M.-P. A cationic NCN pincer Platinum(II) aquo complex with a bis(imidazolinyl) phenyl ligand: Studies toward its synthesis and asymmetric Friedel–Crafts alkylation of indoles with nitroalkenes. Organometallics 2012, 31, 835–846. [Google Scholar] [CrossRef]
- Wu, L.-Y.; Hao, X.-Q.; Xu, Y.-X.; Jia, M.-Q.; Wang, Y.-N.; Gong, J.-F.; Song, M.-P. Chiral NCN pincer Pt(II) and Pd(II) complexes with 1,3-bis(2′-imidazolinyl) benzene: Synthesis via direct metalation, characterization, and catalytic activity in the friedel−crafts alkylation reaction. Organometallics 2009, 28, 3369–3380. [Google Scholar] [CrossRef]
- Schinnerl, M.; Seitz, M.; Kaiser, A.; Reiser, O. New applications of bis(oxazoline) ligands in catalysis: Asymmetric 1,2 and 1,4 addition of znr2 to carbonyl compounds. Org. Lett. 2001, 3, 4259–4262. [Google Scholar] [CrossRef] [PubMed]
- Shintani, R.; Fu, G.C. Copper-catalyzed enantioselective conjugate addition of diethylzinc to acyclic enones in the presence of planar-chiral phosphaferrocene-oxazoline ligands. Org. Lett. 2002, 4, 3699–3702. [Google Scholar] [CrossRef] [PubMed]
- Al Majid, A.M.; Islam, M.S.; Al-Othman, Z.A.; Al-Salhoob, A.F. Enantioselective additions of diethylzinc to aldehydes catalyzed by titanate(IV) complex with chiral bidentate bis-amide ligands based on cyclopropane backbone. Arab. J. Chem. 2017, 10, S964–S970. [Google Scholar] [CrossRef]
- Mei, L.-y.; Yuan, Z.-l.; Shi, M. Chiral imidazoline–phosphine ligands for palladium-catalyzed asymmetric allylic substitutions. Organometallics 2011, 30, 6466–6475. [Google Scholar] [CrossRef]
- Dugal-Tessier, J.; Dake, G.R.; Gates, D.P. Chiral phosphaalkene− oxazoline ligands for the palladium-catalyzed asymmetric allylic alkylation. Org. Lett. 2010, 12, 4667–4669. [Google Scholar] [CrossRef]
- Sawada, T.; Nakada, M. Preparation of new chiral bisoxazoline ligands for the catalytic asymmetric intramolecular cyclopropanation of α-diazo-β-keto phenyl sulfone to afford a useful bicyclo [3.1.0] hexane derivative. Tetrahedron Asymmetry 2012, 23, 350–356. [Google Scholar] [CrossRef]
- Burguete, M.I.; Fraile, J.M.; García, J.I.; García-Verdugo, E.; Herrerías, C.I.; Luis, S.V.; Mayoral, J.A. Bis(oxazoline) copper complexes covalently bonded to insoluble support as catalysts in cyclopropanation reactions. J. Org. Chem. 2001, 66, 8893–8901. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.-X.; Zhu, S.-F.; Yang, Y.; Zhou, Q.-L. Asymmetric friedel− crafts alkylations of indoles with nitroalkenes catalyzed by zn(ii)−bisoxazoline complexes. J. Org. Chem. 2006, 71, 75–80. [Google Scholar] [CrossRef]
- Itoh, J.; Fuchibe, K.; Akiyama, T. Chiral phosphoric acid catalyzed enantioselective Friedel–Crafts alkylation of indoles with nitroalkenes: Cooperative effect of 3Å molecular sieves. Angew. Chem. Int. Ed. 2008, 47, 4016–4018. [Google Scholar] [CrossRef]
- Ganesh, M.; Seidel, D. Catalytic enantioselective additions of indoles to nitroalkenes. J. Am. Chem. Soc. 2008, 130, 16464–16465. [Google Scholar] [CrossRef]
- Liu, H.; Du, D.M. Development of diphenylamine-linked bis(imidazoline) ligands and their application in asymmetric Friedel–Crafts alkylation of indole derivatives with nitroalkenes. Adv. Synth. Catal. 2010, 352, 1113–1118. [Google Scholar] [CrossRef]
- Buchcic, A.; Zawisza, A.; Leśniak, S.; Rachwalski, M. Asymmetric Friedel–Crafts alkylation of indoles catalyzed by chiral aziridine-phosphines. Catalysts 2020, 10, 971. [Google Scholar] [CrossRef]
- Gao, M.-Z.; Wang, B.; Liu, H.-B.; Xu, Z.-L. Synthesis of chiral 2,5-bis(oxazolinyl)thiophenes and their application as chiral shift reagents for 1,1′-bi-2-naphthol. Chin. J. Chem. 2002, 20, 85–89. [Google Scholar] [CrossRef]
- Yuan, Z.-L.; Lei, Z.-Y.; Shi, M. Binam and h8-binam-based chiral imines and Zn(OTf)2 catalyzed enantioselective Friedel–Crafts alkylation of indoles with nitroalkenes. Tetrahedron Asymmetry 2008, 19, 1339–1346. [Google Scholar] [CrossRef]
- Meshram, H.M.; Kumar, D.A.; Reddy, B.C. Simple and efficient Friedel–Crafts alkylation of 1h-indole with electron-deficient alkenes promoted by zinc acetate. Helv. Chim. Acta 2009, 92, 1002–1006. [Google Scholar] [CrossRef]
- Yongcheng, C.; Yuanyuan, C.; Zhengfeng, X.; Wenping, W. Friedel–Crafts reaction of indoles with nitroalkenes catalyzed by Yb(OTf)3. Chinese J. Org. Chem. 2011, 31, 1672–1677. [Google Scholar]
- Liang, L.; Liu, Q.; Zhang, J.; Wang, F.; Yuan, Y. Efficient iron-catalyzed michael addition of indole to nitroolefins under solvent-free conditions. Res. Chem. Intermed. 2013, 39, 1957–1962. [Google Scholar] [CrossRef]
- Jalal, S.; Sarkar, S.; Bera, K.; Maiti, S.; Jana, U. Synthesis of nitroalkenes involving a cooperative catalytic action of iron(III) and piperidine: A one-pot synthetic strategy to 3-alkylindoles, 2h-chromenes and n-arylpyrrole. Eur. J. Org. Chem. 2013, 2013, 4823–4828. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry [a] | Ligands | L:Cu(OTf)2 [1:1] | Solvents | Time [h] | Yield (%) [b] | ee (%) [c,d] |
|---|---|---|---|---|---|---|
| 1. | L1 | 15 mol% | Toluene | 48 | 78 | 5 |
| 2. | L2 | 15 mol% | Toluene | 48 | 75 | 3 |
| 3. | L3 | 15 mol% | Toluene | 48 | 77 | 3 |
| 4. | L4 | 15 mol% | Toluene | 48 | 70 | 45 |
| 5. | L5/, | 15 mol% | Toluene | 48 | 66 | 75 |
| 6. | L5 | 15 mol% | Toluene | 72 | 68 | 74 |
| 7. | L5 | 5 mol% | Toluene | 48 | 20 | 65 |
| 8. | L5 | 10 mol% | Toluene | 48 | 46 | 71 |
| 9. | L5 | 20 mol% | Toluene | 48 | 65 | 74 |
| 10. | L5 | 15 mol% | THF | 48 | 55 | 50 |
| 11. | L5 | 15 mol% | MeOH | 72 | 30 | 5 |
| 12. | L5 | 15 mol% | ACN | 96 | 10 | 4 |
| 13. | L5 | 15 mol% | DCM | 72 | 80 | 0 |
| 14. | L5 | 15 mol% | Hexane | 72 | - | - |
| 15. | L5 | 15 mol% | EA | 96 | traces | - |

| Entry [a] | Metals Salts (15 mol%) | Time [h] | Temp [°C] | Yield (%) [b] | ee (%) [c,d] |
|---|---|---|---|---|---|
| 1. | Zn(OTf)2 | 48 | 25 | 97 | 10 |
| 2. | Mg(OTf)2 | 72 | 25 | - | - |
| 3. | Er(OTf)2 | 72 | 25 | 40 | 2 |
| 4. | Yb(OTf)2 | 72 | 25 | 47 | 0 |
| 5. | FeCl3 | 24 | 25 | 80 | 2 |
| 6. | PdCl2 | 24 | 25 | 70 | 0 |
| 7. | Cu(OTf)2 | 92 | 0 | 42 | 76 |
| 8. | Cu(OTf)2 | 24 | 70 | 66 | 65 |

| Entry [a] | R1 (9a–h) | R2 | R3 | 10a–i | Yields (%) [b] | ee (%) [c] | R/S | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1. | 4-F-C6H4 | H | H | 10a | 67 76[LS] | 74 77[LS] | (S) [d] | [74] |
| 2. | 3-Br-C6H4 | H | H | 10b | 64 | 80 | (S) [d] | [74,76] |
| 3. | 4-CF3-C6H4 | H | H | 10c | 40 | 75 | (S) [d] | [75] |
| 4. | 4-CH3O-C6H4 | H | H | 10d | 66 | 69 | (S) [d] | [74] |
| 5. | 2-NO2-C6H4 | H | H | 10e | 58 | 70 | (R) [d] | [80] |
| 6. | 2,4-Cl2-C6H3 | H | H | 10f | 48 | 71 | (R) [d] | [74] |
| 7. | 2-thienyl | H | H | 10g | 52 | 71 | (S) [e] | [42] |
| 8. | 2,6-Cl2-C6H3 | H | H | 10h | 60 | 64 | (R) [e] | [41] |
| 9. | 4-F-C6H4 | Br | H | 10i | 55 | 77 | (S) [e] | |
| 10. | 3-Br-C6H4 | Br | H | 10j | 46 | 81 | (S) [e] | |
| 11. | 4-CF3-C6H4 | Br | H | 10k | 35 | 79 | (S) [e] | |
| 12. | 4-CH3O-C6H4 | Br | H | 10l | 39 | 63 | (S) [d] | [81] |
| 13. | 2-NO2-C6H4 | Br | H | 10m | 42 | 78 | (R) [e] | |
| 14. | 2,4-Cl2-C6H3 | Br | H | 10n | 37 | 75 | (R) [e] | |
| 15. | 2-thienyl | Br | H | 10o | 47 | 72 | (S) [e] | [42] |
| 16. | 2,6-Cl2-C6H3 | Br | H | 10p | 52 | 60 | (R) [e] | |
| 17. | 2-thienyl | F | H | 10q | 57 | 66 | (S) [e] | |
| 18. | 2,6-Cl2-C6H3 | F | H | 10r | 45 | 21 | (R) [e] | |
| 19. | 4-F-C6H4 | H | Et | 10s | 73 | 35 | (S) [e] | |
| 20. | 4-CH3O-C6H4 | H | Et | 10t | 76 | 27 | (S) [e] | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.S.; Alammari, A.S.; Barakat, A.; Alshahrani, S.; Haukka, M.; Al-Majid, A.M. Exploiting the Chiral Ligands of Bis(imidazolinyl)- and Bis(oxazolinyl)thiophenes—Synthesis and Application in Cu-Catalyzed Friedel–Crafts Asymmetric Alkylation. Molecules 2021, 26, 7408. https://doi.org/10.3390/molecules26237408
Islam MS, Alammari AS, Barakat A, Alshahrani S, Haukka M, Al-Majid AM. Exploiting the Chiral Ligands of Bis(imidazolinyl)- and Bis(oxazolinyl)thiophenes—Synthesis and Application in Cu-Catalyzed Friedel–Crafts Asymmetric Alkylation. Molecules. 2021; 26(23):7408. https://doi.org/10.3390/molecules26237408
Chicago/Turabian StyleIslam, Mohammad Shahidul, Abdullah Saleh Alammari, Assem Barakat, Saeed Alshahrani, Matti Haukka, and Abdullah Mohammed Al-Majid. 2021. "Exploiting the Chiral Ligands of Bis(imidazolinyl)- and Bis(oxazolinyl)thiophenes—Synthesis and Application in Cu-Catalyzed Friedel–Crafts Asymmetric Alkylation" Molecules 26, no. 23: 7408. https://doi.org/10.3390/molecules26237408
APA StyleIslam, M. S., Alammari, A. S., Barakat, A., Alshahrani, S., Haukka, M., & Al-Majid, A. M. (2021). Exploiting the Chiral Ligands of Bis(imidazolinyl)- and Bis(oxazolinyl)thiophenes—Synthesis and Application in Cu-Catalyzed Friedel–Crafts Asymmetric Alkylation. Molecules, 26(23), 7408. https://doi.org/10.3390/molecules26237408