
BiLSTM-5mC: A Bidirectional Long Short-Term Memory-Based Approach for Predicting 5-Methylcytosine Sites in Genome-Wide DNA Promoters


Abstract
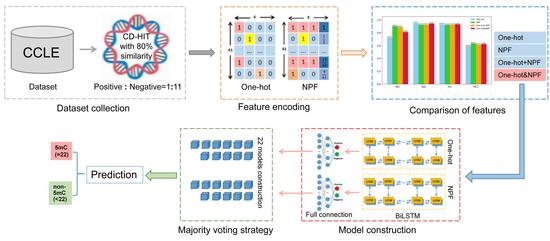
:
1. Introduction
2. Results and Discussions
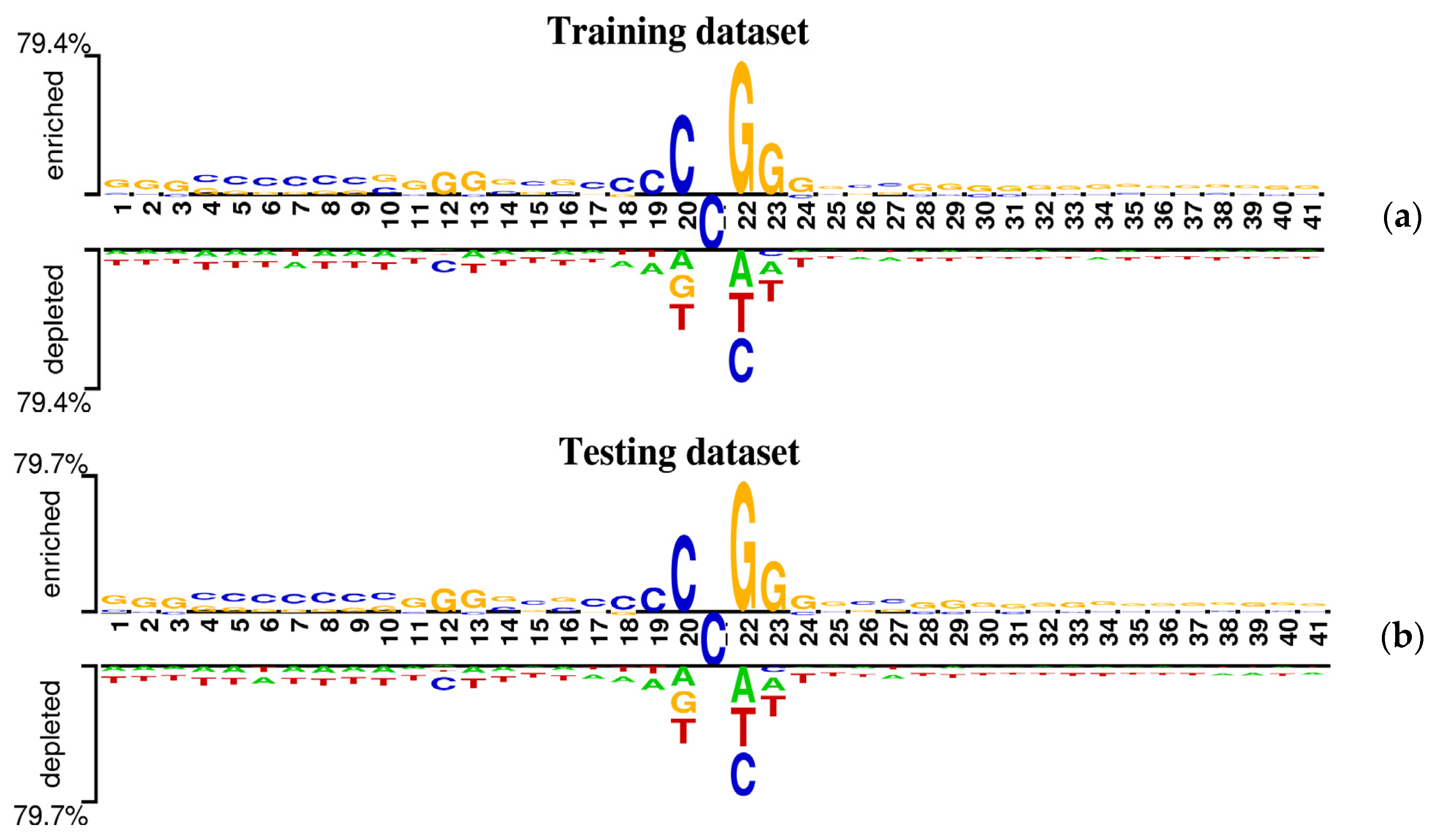
2.1. Sequence Composition Analysis
2.2. Performance Evaluation on Different Feature Encoding Methods
2.3. Performance Comparison with Existing Methods
3. Materials and Methods
3.1. Benchmark Datasets
3.2. Feature Encoding Schemes
3.2.1. One-Hot Encoding
3.2.2. The Nucleotide Property and Frequency
3.3. Model Construction
3.3.1. The Overall Framework
3.3.2. Bidirectional Long Short-Term Memory Network
3.3.3. Fully Connected Network
3.4. Performance Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Lv, H.; Dao, F.Y.; Zhang, D.; Yang, H.; Lin, H. Advances in mapping the epigenetic modifications of 5-methylcytosine (5mC), N6-methyladenine (6mA), and N4-methylcytosine (4mC). Biotechnol. Bioeng. 2021, 118, 4204–4216. [Google Scholar] [CrossRef]
- Michalak, E.; Burr, M.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Javierre, B.M.; Fernandez, A.F.; Richter, J.; Al-Shahrour, F.; Martin-Subero, J.I.; Rodriguez-Ubreva, J.; Bersadco, M.; Fraga, M.F.; O’Hanlon, T.P.; Rider, L.G.; et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ubreva, J.; de la Calle-Fabregat, C.; Li, T.; Ballestar, M.L.; Català-Moll, F.; Morante-Palacios, O.; Garcia-Gomez, A.; Ceils, R.; Humby, F.; Nerviani, A.; et al. Inflammatory cytokines shape a changing DNA methylome in monocytes mirroring disease activity in rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Sawalha, A.H.; Lu, Q. Clinical value of DNA methylation markers in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 514–524. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Joosten, S.C.; Feng, Z.; De Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Van Tilborg, A.A.; Zwarthoff, E.C. DNA methylation-based biomarkers in bladder cancer. Nat. Rev. Urol. 2013, 10, 327–335. [Google Scholar] [CrossRef]
- Frigola, J.; Solé, X.; Paz, M.F.; Moreno, V.; Esteller, M.; Capellà, G.; Peinado, M.A. Differential DNA hypermethylation and hypomethylation signatures in colorectal cancer. Hum. Mol. Genet. 2004, 14, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Murphy, R.F.; Agrawal, D.K. DNA methylation in breast and colorectal cancers. Mod. Pathol. 2007, 20, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tollefsbol, T.O. DNA Methylation Detection: Bisulfite Genomic Sequencing Analysis. Methods Mol. Biol. 2011, 791, 11–21. [Google Scholar]
- Booth, M.; Ost, T.W.B.; Beraldi, D.; Bell, N.M.; Branco, M.; Reik, W.; Balasubramanian, S. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat. Protoc. 2013, 8, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Siejka-Zielińska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Böckler, B.; Song, C.-X. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Cairns, B.R. Transcriptome-wide target profiling of RNA cytosine methyltransferases using the mechanism-based enrichment procedure Aza-IP. Nat. Protoc. 2014, 9, 337–361. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, Z.-M.; Li, S.-H.; Tan, J.-X.; Chen, W.; Lin, H. Evaluation of different computational methods on 5-methylcytosine sites identification. Briefings Bioinform. 2019, 21, 982–995. [Google Scholar] [CrossRef]
- Bhasin, M.; Zhang, H.; Reinherz, E.L.; Reche, P. Prediction of methylated CpGs in DNA sequences using a support vector machine. FEBS Lett. 2005, 579, 4302–4308. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Fan, S.; Zhang, X.; Zhang, M.Q. Predicting methylation status of CpG islands in the human brain. Bioinformatics 2006, 22, 2204–2209. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Qiu, W.R.; Chou, K.C. iDNA-Methyl: Identifying DNA methylation sites via pseudo trinucleotide composition. Anal. Biochem. 2015, 474, 69–77. [Google Scholar] [CrossRef]
- Amoreira, C.; Hindermann, W.; Grunau, C. An improved version of the DNA methylation database (MethDB). Nucleic Acids Res. 2003, 31, 75–77. [Google Scholar] [CrossRef]
- Li, J.; Huang, Y.; Yang, X.; Zhou, Y.; Zhou, Y. RNAm5Cfinder: A Web-server for Predicting RNA 5-methylcytosine (m5C) Sites Based on Random Forest. Sci. Rep. 2018, 8, 17299. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.R.; Jiang, S.Y.; Xu, Z.C.; Xiao, X.; Chou, K.C. iRNAm5C-PseDNC: Identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget 2017, 8, 41178–41188. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Zhang, Z.; Sun, R.; Zhu, L.; He, J.; Huang, B.; Xiong, Y.; Zhu, X. RNAm5CPred: Prediction of RNA 5-Methylcytosine Sites Based on Three Different Kinds of Nucleotide Composition. Mol. Ther. Nucleic Acids 2019, 18, 739–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, S.; Hayat, M.; Iqbal, M.; Tahir, M. iRNA-PseTNC: Identification of RNA 5-methylcytosine sites using hybrid vector space of pseudo nucleotide composition. Front. Comput. Sci. 2020, 14, 451–460. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, Y.; Liu, Y.; Chen, Y.; Bi, S.; Zhu, X. m5CPred-SVM: A novel method for predicting m5C sites of RNA. BMC Bioinform. 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Li, X.; Ding, H.; Xu, L.; Xiang, H. Prediction of m5C Modifications in RNA Sequences by Combining Multiple Sequence Features. Mol. Ther.-Nucleic Acids 2020, 21, 332–342. [Google Scholar] [CrossRef]
- Feng, P.; Ding, H.; Chen, W.; Lin, H. Identifying RNA 5-methylcytosine sites via pseudo nucleotide compositions. Mol. BioSyst. 2016, 12, 3307–3311. [Google Scholar] [CrossRef]
- Sabooh, M.F.; Iqbal, N.; Khan, M.; Khan, M.; Maqbool, H.F. Identifying 5-methylcytosine sites in RNA sequence using composite encoding feature into Chou’s PseKNC. J. Theor. Biol. 2018, 452, 1–9. [Google Scholar] [CrossRef]
- Song, J.; Zhai, J.; Bian, E.; Song, Y.; Yu, J.; Ma, C. Corrigendum: Transcriptome-Wide Annotation of m5C RNA Modifications Using Machine Learning. Front. Plant Sci. 2018, 9, 1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Xu, Y.; Li, L.; Liu, Z.; Yang, X.; Yu, D.-J. Accurate RNA 5-methylcytosine site prediction based on heuristic physical-chemical properties reduction and classifier ensemble. Anal. Biochem. 2018, 550, 41–48. [Google Scholar] [CrossRef]
- Li, J.; Huang, Y.; Zhou, Y. A Mini-review of the Computational Methods Used in Identifying RNA 5-Methylcytosine Sites. Curr. Genom. 2020, 21, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Li, H.; Ning, S.; Ghandi, M.; Kryukov, G.; Gopal, S.; Deik, A.; Souza, A.; Pierce, K.; Keskula, P.; Hernandez, D.; et al. The landscape of cancer cell line metabolism. Nat. Med. 2019, 25, 850–860. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, X.; Xu, Z.-C. iPromoter-5mC: A Novel Fusion Decision Predictor for the Identification of 5-Methylcytosine Sites in Genome-Wide DNA Promoters. Front. Cell Dev. Biol. 2020, 8, 614. [Google Scholar] [CrossRef]
- Nguyen, D.; Tran, T.-A.; Khanh, L.N.Q.; Pham, D.-M.; Ou, Y.-Y. An extensive examination of discovering 5-Methylcytosine Sites in Genome-Wide DNA Promoters using machine learning based approaches. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Tang, H.; Ding, H.; Lin, H. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics 2016, 107, 255–258. [Google Scholar] [CrossRef]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-methyladenosine Sites Using Ensemble Learning. Mol. Ther.-Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, F.; Majumdar, S.; Darabi, H.; Chen, S. LSTM Fully Convolutional Networks for Time Series Classification. IEEE Access 2017, 6, 1662–1669. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Xu, J.; Wang, X.; Peng, X.; Song, J.; Yu, D.-J. Leveraging the attention mechanism to improve the identification of DNA N6-methyladenine sites. Briefings Bioinform. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Ning, Q.; Sheng, M. m7G-DLSTM: Intergrating directional Double-LSTM and fully connected network for RNA N7-methlguanosine sites prediction in human. Chemom. Intell. Lab. Syst. 2021, 217, 104398. [Google Scholar] [CrossRef]
- Jurtz, V.I.; Johansen, A.R.; Nielsen, M.; Armenteros, J.J.A.; Nielsen, H.; Sønderby, C.K.; Winther, O.; Sønderby, S.K. An introduction to deep learning on biological sequence data: Examples and solutions. Bioinformatics 2017, 33, 3685–3690. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Feature | Algorithm |
|---|---|---|
| iPromoter-5mC [36] | One-hot | Deep neural network |
| 5mC_Pred [37] | K-mers | XGBoost |
| BiLSTM-5mC (This study) | One-hot and NPF | BiLSTM |
| Method | Sen | Spe | Acc | MCC | AUC |
|---|---|---|---|---|---|
| iPromoter-5mC | 0.8746 | 0.9039 | 0.9016 | 0.5743 | 0.9566 |
| 5mC_Pred | 0.8990 | 0.9200 | 0.9180 | 0.6260 | 0.9620 |
| BiLSTM-5mC | 0.8096 | 0.9404 | 0.9302 | 0.6235 | 0.9644 |
| Method | Sen | Spe | Acc | MCC | AUC |
|---|---|---|---|---|---|
| iPromoter-5mC | 0.8777 | 0.9042 | 0.9022 | 0.5771 | 0.9570 |
| 5mC_Pred | 0.8950 | 0.9200 | 0.9180 | 0.6250 | 0.9620 |
| BiLSTM-5mC | 0.8661 | 0.9374 | 0.9303 | 0.6384 | 0.9635 |
| Dataset | Positive Sample | Negative Sample |
|---|---|---|
| Training dataset | 55,800 | 658,861 |
| Testing dataset | 13,950 | 164,715 |
| Total | 69,750 | 823,576 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Wang, J.; Li, Q.; Liu, T. BiLSTM-5mC: A Bidirectional Long Short-Term Memory-Based Approach for Predicting 5-Methylcytosine Sites in Genome-Wide DNA Promoters. Molecules 2021, 26, 7414. https://doi.org/10.3390/molecules26247414
Cheng X, Wang J, Li Q, Liu T. BiLSTM-5mC: A Bidirectional Long Short-Term Memory-Based Approach for Predicting 5-Methylcytosine Sites in Genome-Wide DNA Promoters. Molecules. 2021; 26(24):7414. https://doi.org/10.3390/molecules26247414
Chicago/Turabian StyleCheng, Xin, Jun Wang, Qianyue Li, and Taigang Liu. 2021. "BiLSTM-5mC: A Bidirectional Long Short-Term Memory-Based Approach for Predicting 5-Methylcytosine Sites in Genome-Wide DNA Promoters" Molecules 26, no. 24: 7414. https://doi.org/10.3390/molecules26247414
APA StyleCheng, X., Wang, J., Li, Q., & Liu, T. (2021). BiLSTM-5mC: A Bidirectional Long Short-Term Memory-Based Approach for Predicting 5-Methylcytosine Sites in Genome-Wide DNA Promoters. Molecules, 26(24), 7414. https://doi.org/10.3390/molecules26247414