3.2.1. 8D Ethene
The initial 8D study of ethene used all normal modes except those which are best described as C-H stretches of various combinations (the highest frequency modes). The symmetry labels (in the D
point group) of the modes are shown in
Table 4 along with their frequencies at the level of electronic structure theory used here.
The 8D dynamics of ethene was performed using the DVR bases shown in
Table 4 along the eight mass-frequency scaled normal modes on all three electronic states. The initial wavefunction was constructed as an 8D Gaussian function of width
, centred at the origin of the coordinate system (the Franck-Condon geometry) and placed on the first excited state (with B
symmetry). The wavepacket was propagated for 100 fs using the default ABM integrator for MCTDH. Four 2D combined modes were used: {1B
,6B
}, {2B
,4B
}, {5A
,7A
}, {3B
,8A
}. 9 SPFs were used for each 2D combined mode to expand the wavefunction on the ground and second excited state (both of A
symmetry) whilst 15 SPFs each were used on the initial state.
To construct the PES, 100 coordinates were sampled every 1 fs around the centre of the wavefunction on each state, and energies added to the database if the KRR variance at the selected geometry exceeded
. Adiabatic energies for all three states were calculated at the SA(3)-CASSCF(2,2)/6-31G(d,p) level before being diabatised using the Procrustes diabatisation scheme. In addition to the KRR variance being used to determine which points to add to the database, an energy threshold of 20 eV was also used, such that if the calculated ground state energy exceeded this value it was rejected. In addition, before carrying out an electronic structure calculation, the KRR predicted energy was evaluated there and if found to be greater than 22 eV (a 10% discrepancy in the threshold to allow for errors in the KRR extrapolation of the PES), the point was rejected before expending time calculating the energy. The KRR fit of the diabatic energies and couplings was achieved using the additive kernel (Equation (
13)) with
along each mode. A total of 493 points were retained in the database compiled over the course of the dynamics. Finally, a secondary SVD fitting of the KRR PES was performed, with enough terms being retained to keep
. The number of terms in this expansion varied during the dynamics, depending on the number of sampled points in the KRR fit, but ranged from an initial 827, up to a maximum of 1421 before ending at 1387.
After propagation, the dynamics on the ground and initial excited states were separately analysed by DM. We used and . Here, 20 gridpoints were sampled every 10 fs and for the ground state analysis, the first sampling time was 10 fs to allow time for the wavepacket to begin transferring from the excited state.
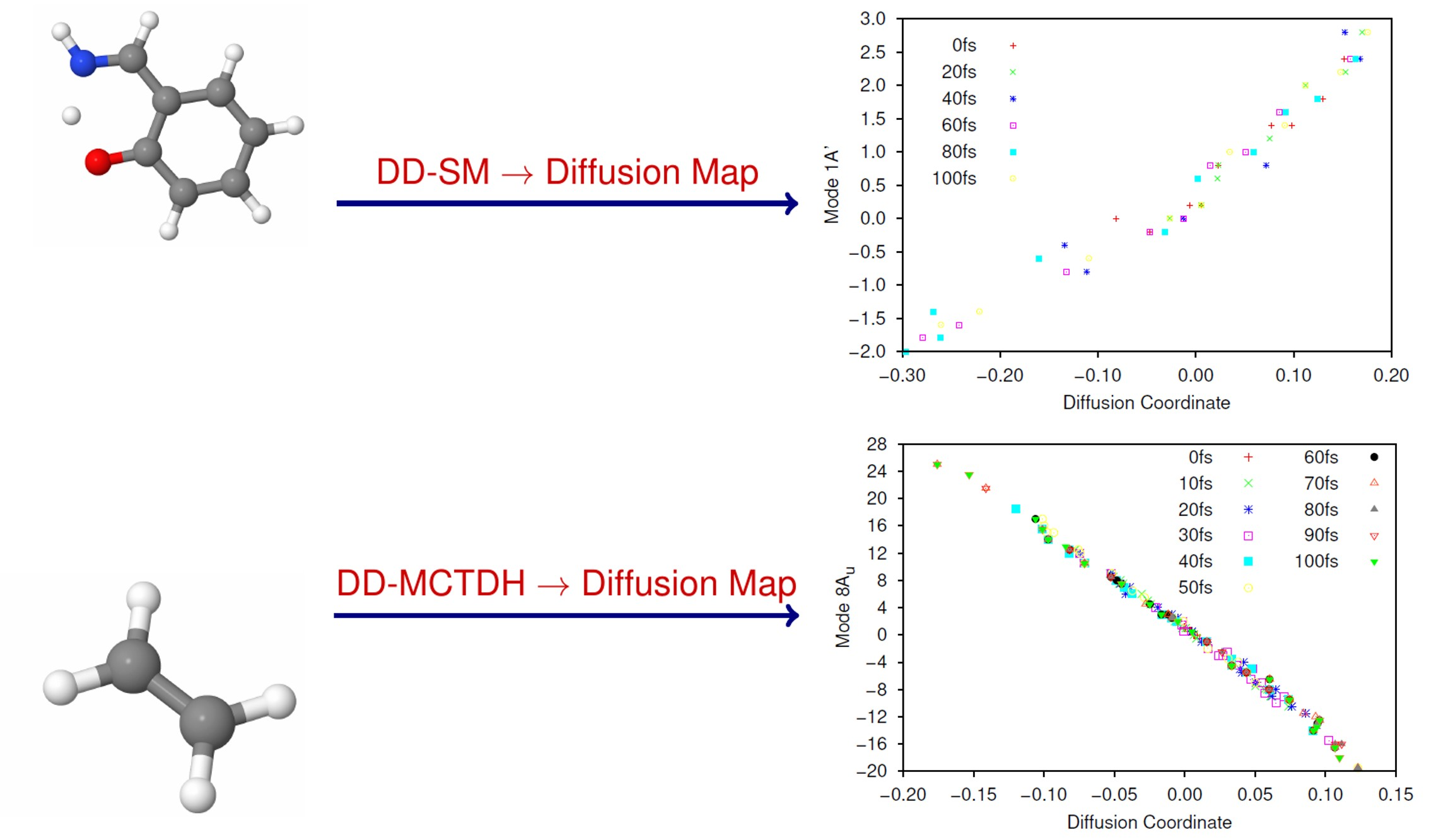
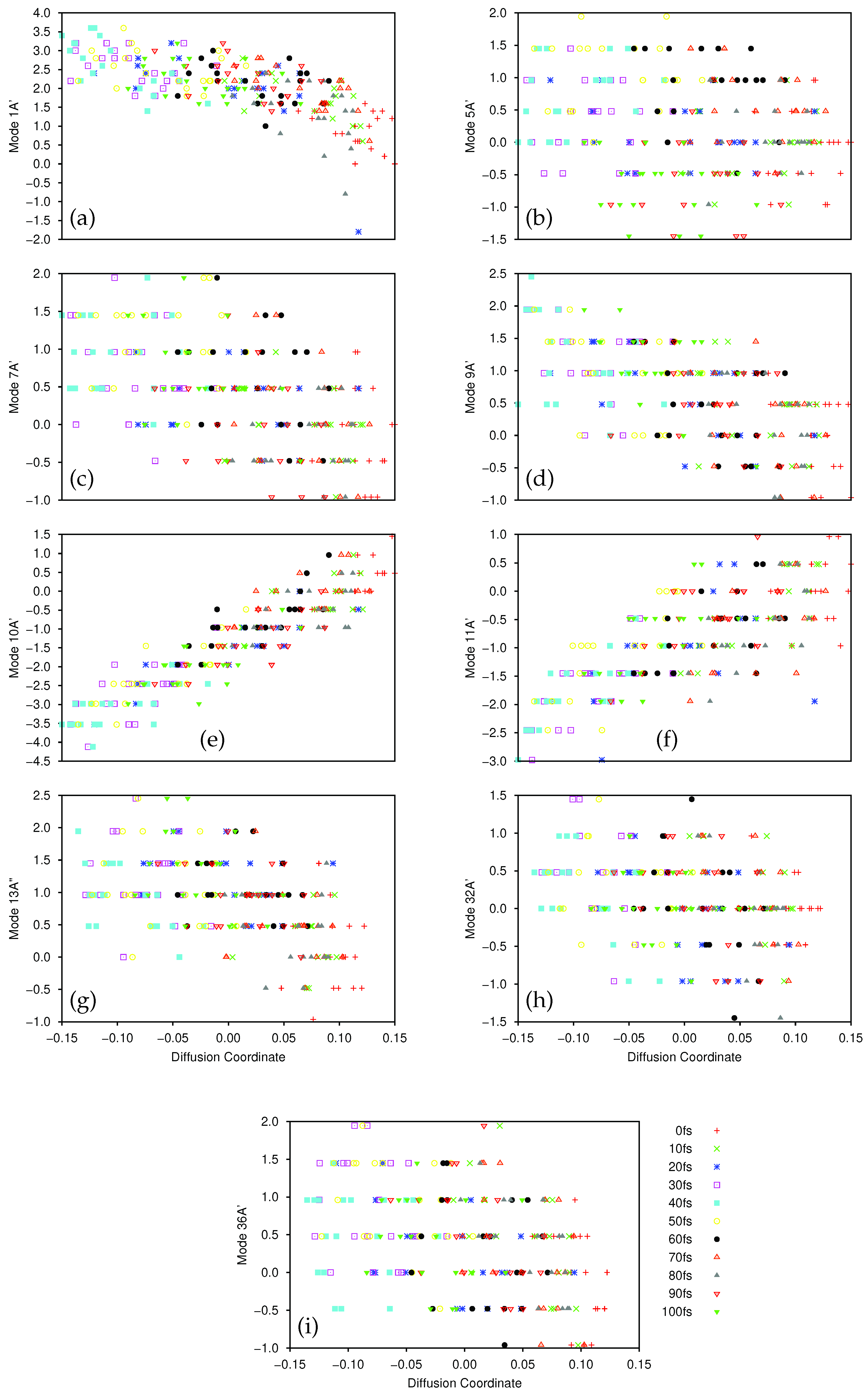
In
Figure 5 and
Figure 6 we present plots of the results of the DM analysis, showing the correlation between the diffusion coordinates on the ground (
Figure 5) and first excited (
Figure 6) electronic states and the coordinates of the associated sampled points along the eight normal modes (mass-frequency scaled). We note that the three highest, non-trivial eigenvalues are: for the ground state, 0.235, 0.106 and 0.0881; for the excited state motion, 0.651, 0.370 and 0.233. Again we see a significant spectral gap between the two largest eigenvalues indicating that the overall motion of ethene in both states, after excitation from and subsequent non-radiative transfer back to the ground state, is dominated by a single diffusion coordinate. Looking at the eight plots in both figures, it is clear that the diffusion coordinate is dominated by the 8A
normal mode (i.e., it correlates best with the diffusion coordinate) on both states; this mode is essentially a torsion mode, with the two CH
groups rotating in opposite directions. All other normal modes show little or no correlation with the diffusion coordinate. Such torsional motion is expected to dominate for ethene, the excitation corresponding to a breaking of the ethene
-bond hence allowing free rotation around the C-C bond i.e., giving a
cis-
trans isomerization motion. So, we see that the diffusion map method has successfully extracted the most important mode from the large number of dimensions being followed in the wavepacket dynamics.
We note that the gradient of the plots containing the 8A
normal mode and the diffusion coordinate differ for both states (
Figure 5h and
Figure 6h), the ground state diffusion coordinate having positive correlation with the 8A
normal mode coordinate while the correlation is negative for the excited state. This is due to the fact that the diffusion coordinate is defined by the elements of an eigenvector of the diffusion operator matrix, but the overall sign of the eigenvectors of linear operators is arbitrary, hence so is the correlation of the diffusion coordinate with the normal mode.
To see the time-dependence of the diffusion maps more clearly, the data in
Figure 5 and
Figure 6 is presented in the
Supplementary Materials with the sampled points from each timestep having their own plot (
Figures S12–S19 in the Supplementary Materials correspond to those in
Figure 5, whilst
Figures S20–S27 therein represent the data from
Figure 6). Considering the eight plots in
Figure 6 (as well as parts (a) of
Figures S20–S27), describing the dynamics on the excited state, we see the compactness of the initial wavefunction (the cluster of red crosses around the origin in all cases) before the wavefunction begins to move. In all cases we see a spreading of the sampled points over time, which begins soon after excitation. In the case of the 8A
mode this spread is in both directions along the line of correlation between the normal mode and diffusion coordinates, whilst for the other modes the spread of the sampled points is less uniform. For the 1B
mode (
Figure 6 and
Figure S20) the sampled points spread along the diffusion coordinate initially before expanding along the normal mode coordinate up to 40 fs before partially contracting back. A similar delay in the spread of the sampled points along the normal mode is seen for mode 6B
(
Figure 6f and
Figure S25), the first major motion being seen after 20 fs, however, in this case we see no contraction at later times. For mode 4B
the initial wavepacket expansion is followed by a definite motion along the normal mode coordinate between 20 and 40 fs; a full dispersion along the diffusion coordinate is only seen after 70 fs. Considering mode 5A
g (
Figure 6e and
Figure S24), the initial spread of the sampled points is dominated that along the normal mode coordinate with the diffusion coordinate spread coming more slowly. For the remaining modes (2B
, 3B
and 7A
) the spreading of the sampled points in the diffusion map appears to occur in both directions simultaneously. Returning to the ground state (
Figure 5 and
Figures S12–S19 in the Supplementary Materials) we see that the distribution of points is wider to begin with because we only sampled points from 10 fs onwards where the wavepacket on the initially populated excited state had already had time to spread. This indicates that population transfer between the diabatic states is occurring over a wide region of space rather than at localised points. In all modes there is a contraction of distribution of the sampled points along the diffusion coordinate up to 30 fs before a rebound to the full range of the diffusion coordinate In mode 8A
the initial spread of the sampled points follows the line of correlation seen in
Figure 5h whereas for the other modes there is no initial correlation. Further on the subject of population transfer, we note that there was relatively little between the diabatic states over the 100 fs course of the dynamics, the vast majority being over the first 5 fs to the both ground and second excited states; at most 1.5% of the wavepacket transferred to the ground state whilst up to 4.2% went to the upper state.
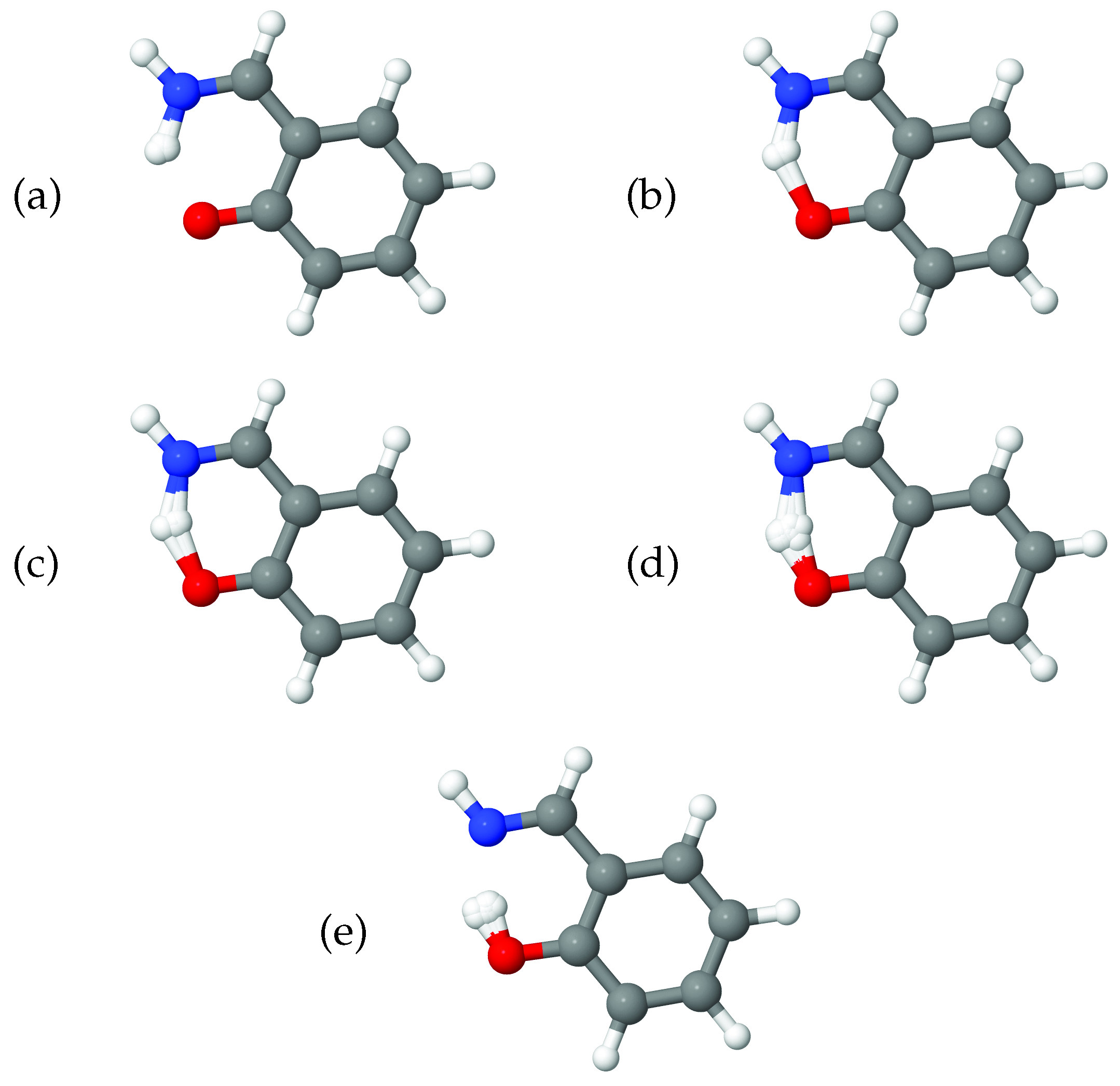
In addition to the plots showing the correlation of the 8A
normal mode with the diffusion coordinate in ethene, in
Figure 7 we show pictures of the ethene geometries, sampled during the diffusion map analysis, grouped according to their diffusion coordinates. In all six diagrams, (i) shows all sampled geometries, superimposed on one another, whilst, for clarity, (ii) gives the average of those geometries. It is apparent that, as we move across the diffusion-coordinate range from
(
Figure 7a) to that of
(
Figure 7f), the angle between the two CH
groups reduces, going through geometries close to planar when
(
Figure 7d) before increasing again in the opposite orientation to that in
Figure 7a. This is further confirmation that the diffusion coordinate can mainly be characterised as a torsion mode, closely related to normal mode 8A
. Each of the panels in
Figure 7 shows many different geometries corresponding to the motion across all normal modes except 8A
; these generate a spread of geometries around the important rotation about the C-C bond. This range corresponds to the uncorrelated spread of geometries sampled along all modes except 8A
, as seen in
Figure 6.
At this point we can compare our results to those of Virshup et al. [
52] where, because they used individual trajectories rather than a grid of basis functions as we are here, they were able to define a diffusion reaction coordinate (DRC) by integrating the Jacobian between the diffusion coordinate and real space coordinates, and then follow selected trajectories along the DRC over time. The previous work by Virshup et al., noted that the pyramidalisation of one of the CH
groups followed the initial torsion motion resulting after electronic excitation; indeed, there is a minimum-energy conical intersection found at such a twisted-pyramidalised geometry resulting from such a torsion-pyramidalisation mechanism. The previously-generated DRC includes this path, and on further to hydrogen migration. In contrast, we see no clear sign of this pyramidalisation in
Figure 5 and
Figure 6, nor in
Figure 7; this is conceivably due to limitations in our coordinate system, the use of normal modes perhaps being too restrictive to observe the pyramidalisation (let alone the hydrogen migration). To further analyse this we can consider the calculated eigenvectors of the diffusion operator with lower eigenvalues i.e.,
,
,
and
. For these eigenvectors we find that
and
are correlated with mode 8A
(albeit more weakly than with
) whilst
and
are weakly correlated with modes 7A
(C-C stretch with hydrogens moving in the opposite direction to their associated carbon) and 5A
(C-C stretch with all hydrogens moving in concert with their bonded carbons), respectively. In none of the five considered eigenvectors do we see correlation with either mode 2B
(out-of-plane motion of all hydrogens in opposite directions at each end of the molecule) or 3B
(out-of-plane bending of all hydrogens in the same direction), which might be considered likely to contribute to the pyramidalisation motion. A different coordinate system may allow such motion to be observed; this conjecture will have to await further implementation to check. As a further check on the dynamics of ethene we also performed calculations in the full-dimensional space of normal modes, which we consider in the next section.
3.2.2. 12D Ethene
Having noted the lack of pyramidalisation in the 8D dynamics of ethene, we finally consider 12D dynamics (i.e., including the four hydrogen stretching modes neglected in
Section 3.2.1) in order to see whether the inclusion of these modes reveals the pyramidalisation.
The same level of electronic structure, and hence optimised geometry and normal modes were used as in the 8D calculations. Information about the DVR bases and mode frequencies are shown in
Table 5. The KRR and SVD sampling and fitting during the dynamics were also as described in
Section 3.2.1, the additional four modes being treated in the same way as the original eight. The initial wavefunction was a Gaussian function of width
in each DOF, centred at the Franck-Condon point on the first excited state. During the course of the dynamics 688 energies were added to the database, and there were a total of 2854 terms in the secondary SVD fit at the end of the wavepacket propagation. The normal modes were combined into six combined modes for the purpose of the time-dependent (SPF) basis, these being: {1B
,2B
}; {3B
,4B
}; {5A
,6B
}; {7A
,10A
}; {8A
}; {9B
,11B
,12B
}. For all combined modes, 5 SPFs were used on the first excited state, whilst a single SPF was used for each of the other two states. A small number of SPFs was necessary to limit memory demands on the computer hardware used, so as a result the propagation is far from converged with respect to the time-dependent basis. It is, however, worth illustrating, with future calculations in mind, whether we can gain useful information about the important DOFs from a poorly converged, high-dimensional calculation, as this will allow subsequent, lower-dimensional calculations to focus on the important modes, and hence to approach convergence.
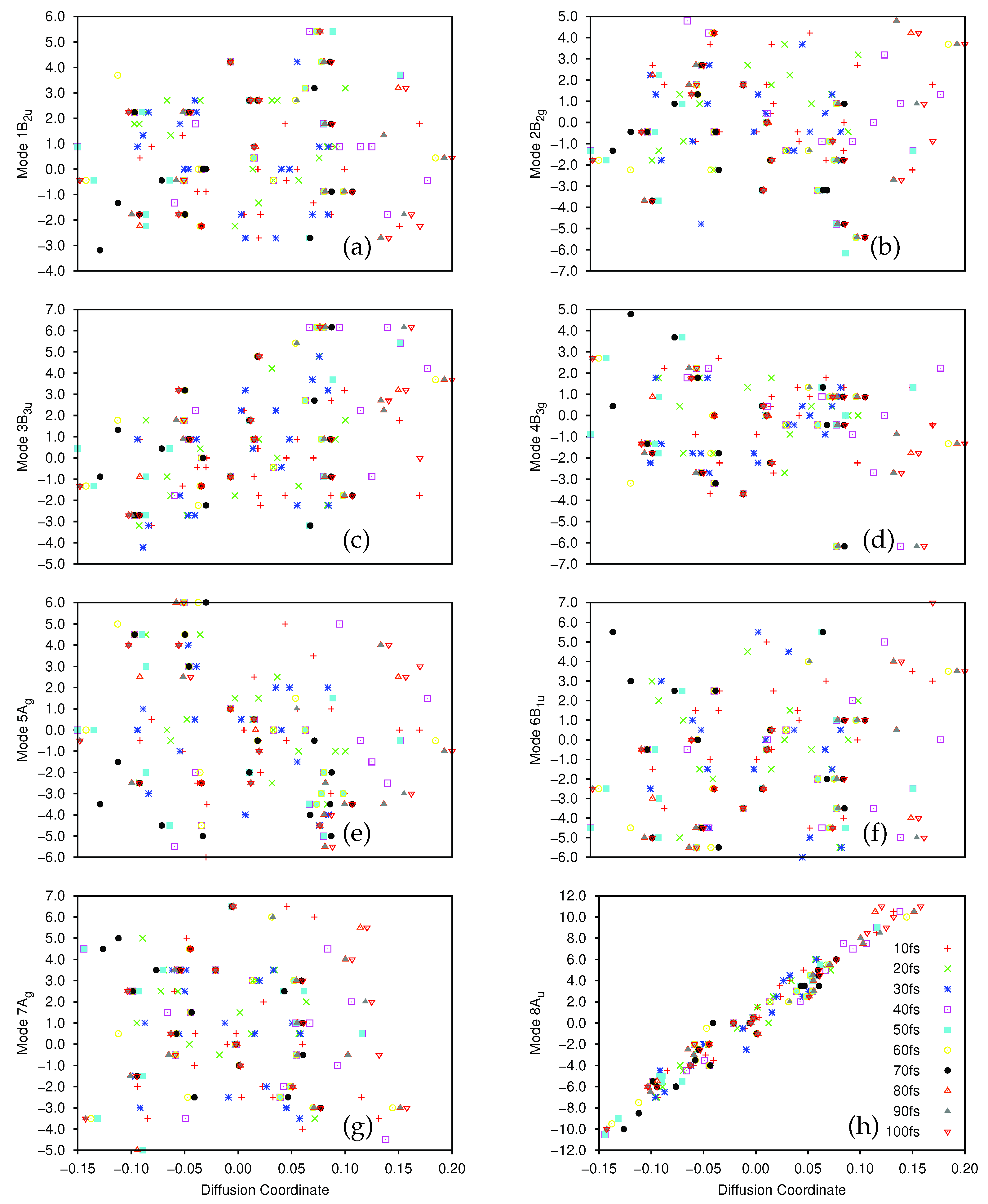
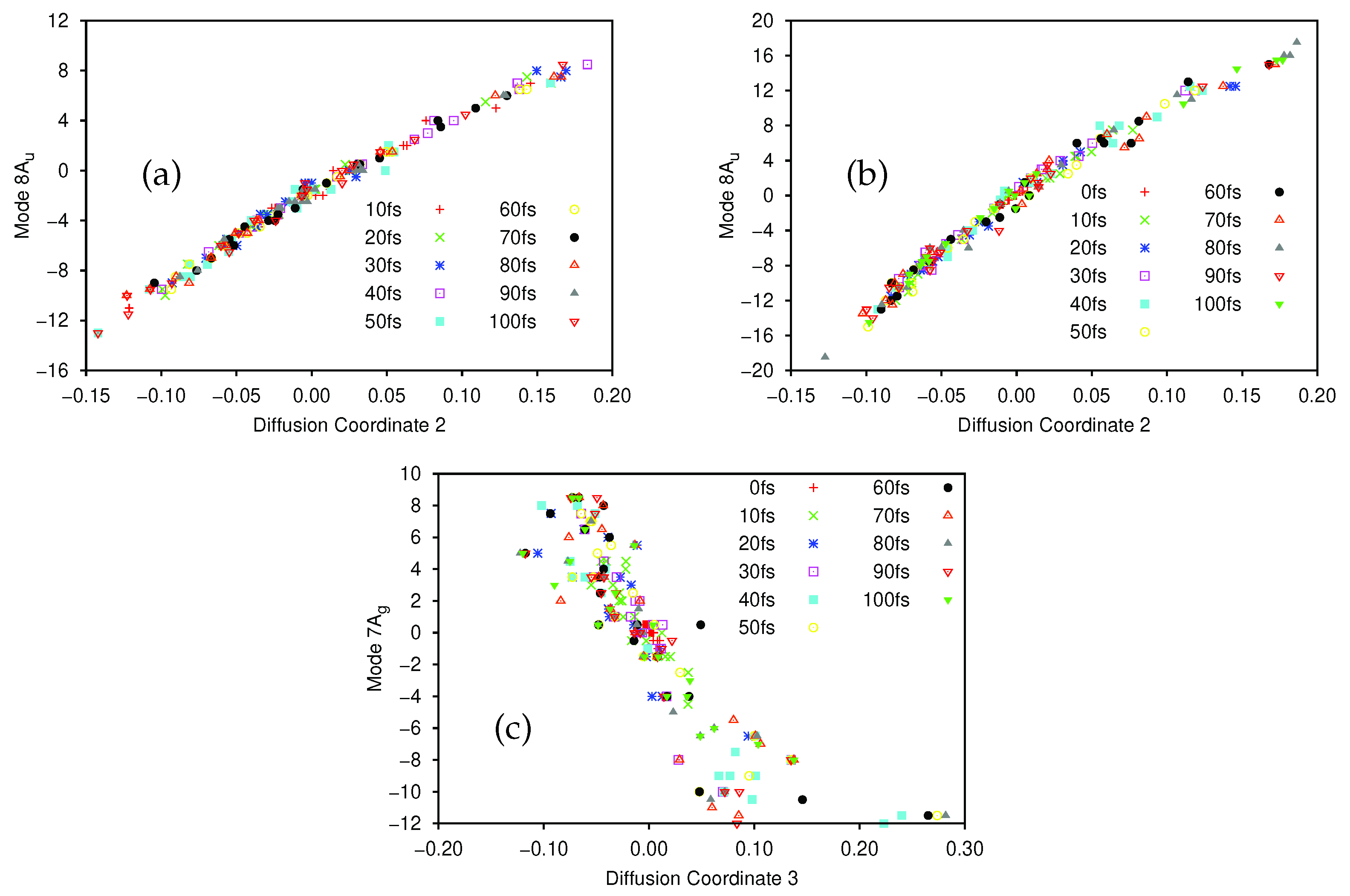
After the dynamics propagation, the wavepacket on the ground and first excited state was analysed using the DM method; 20 points were sampled every 10 fs (the first 10 fs were skipped for the ground state to allow time for population transfer), and we used for the first excited state, for the ground-state, and in all cases. The largest five non-trivial eigenvalues of the diffusion operator were, for the ground state, 0.420, 0.232, 0.180, 0.162 and 0.152, while for the excited state they were 0.480, 0.322, 0.214, 0.204 and 0.154.
There is a clear spectral gap for the ground state, indicating that the dynamics on that state can be well represented by a single diffusion coordinate
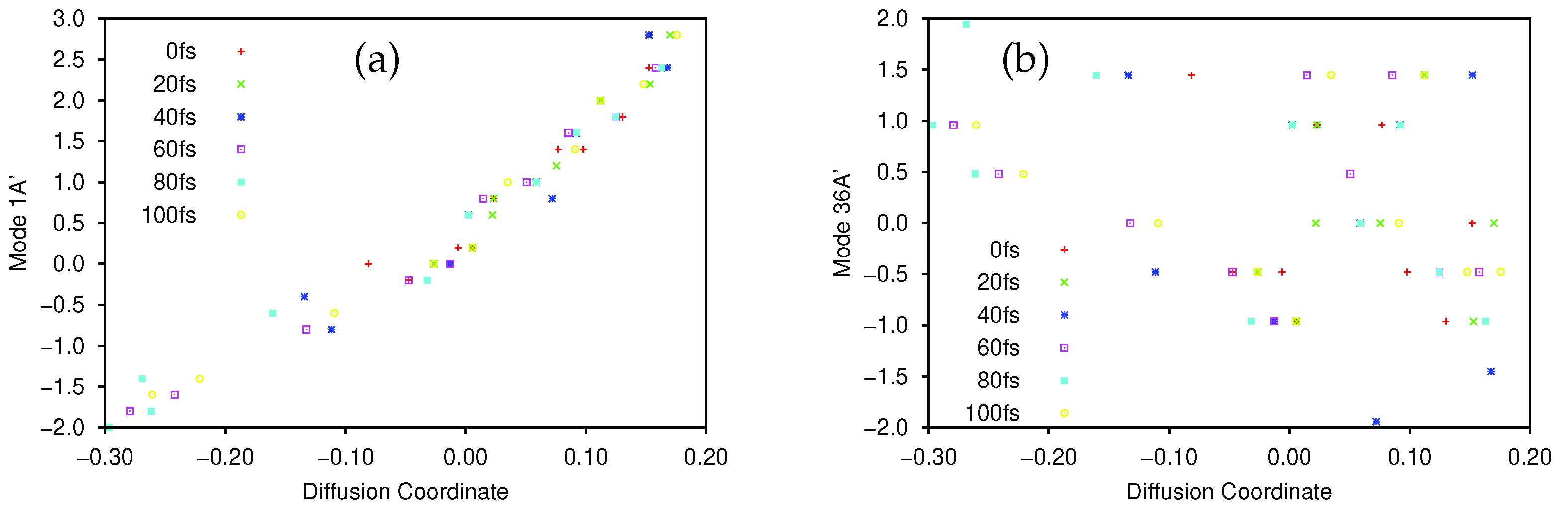
. In
Figure 8a we show the correlation between the sampled 8A
normal mode coordinates and their corresponding diffusion coordinates. The spectral gap is less clear in the excited state case, where the
eigenvector appears to play a more significant role in the dynamics. In
Figure 8b we show the plot of the relationship between the sampled 8A
normal mode coordinates and the
coordinate on the excited state; again we see a very clear correlation between the two. In
Figure 8c we show the correlation between the 7A
mode and
(which is even clearer if the points with
are ignored. As noted in
Section 3.2.1, 7A
is a C-C stretching mode, so even here we see that there is little sign of pyramidalisation. Looking further at
(mixture of 8A
and 10A
), and
and
(both 8A
) on the excited state we see no clear evidence of pyramidalisation whilst, on the ground state, the lower eigenvalue diffusion coordinates correlate with 11B
(
), 8A
/9B
(
), 12B
(
) and 10A
(
) (i.e., all four hydrogen stretching modes are involved along with the torsion, rather than any other out-of-plane motion). It is clear, however, that even with a few SPFs, using all 12 normal modes of ethene, we see the same torsion mode dominating the dynamics as with the better converged 8D calculation; the diffusion map can thus be used to glean useful information.
As with all previous calculations we provide, in the
Supplementary Materials, deconstructed versions of the plots in
Figure 8 (
Figures S28–S30 corresponding to
Figure 8a–c respectively) to help clarify the time evolution of the diffusion map. In much the same way as was seen for the 8D calculation, the sampled points on the excited state (
Figure 8b,c, and
Figures S29 and S30) are initially tightly clustered around the origin, before they spread out along the lines of correlation between the normal mode and diffusion coordinates. On the ground state (
Figure 8a and
Figure S28) the initial set of sampled points is already well distributed, reflecting the fact that the initial sampling was at 10 fs, after the wavepacket had had time to evolve away from the Franck-Condon point. Of potential interest are the small clusters of points locted at the top right of the plots, most easily seen in
Figure S28 at times of 20, 30, 50, 60, 80 and 90 fs, which suggests a splitting of the wavepacket along the torsion mode, although the splitting is less clear at intermediate times.
Taking the analysis further, we performed a calculation to locate the twisted-pyramidalised conical intersection (TPCI) between the ground and first excited states, before using the
vctrans program in the
Quantics package to transform the Cartesian coordinates of the TPCI into the space of the mass-frequency scaled normal modes used for the dynamics. The results of this calculation are shown in
Table 6, from which it is clear that the largest magnitude components are in the C-H stretching modes (9B
, 10A
, 11B
and 12B
), the largest distortion outside these four modes being along the 8A
torsion mode, whose importance is thus confirmed. We saw that the one of the four C-H stretching modes (10A
) made an appearance in the
diffusion coordinate on the excited state, whilst all four appeared in the lower eigenvalue diffusion coordinates on the ground state. Therefore, whilst the dominant diffusion coordinates in the dynamics corresponded to the torsion mode, there is evidence of motion in the C-H stretching modes, which are important to describe the pyramidalisation, in the less significant diffusion coordinates.
A shortcoming of our initial dynamics calculation is also apparent by comparing
Table 5 and
Table 6, where we note that the TPCI coordinate is not actually included in the domain of the DVR grid with the grids along modes 9B
, 11B
and 12B
all being too short. As such a second DD-MCTDH calculation was performed using longer, but less dense for reasons of computational cost, DVR grids which are given in
Table 7 with the frequencies being the same as those in
Table 5. The initial conditions and KRR sampling for the calculation were as for the initial calculation, but the calculation was only run for 90 fs. 665 points were added to the energy database whilst the number of terms in the SVD fit of the PES was 3417 at the end.
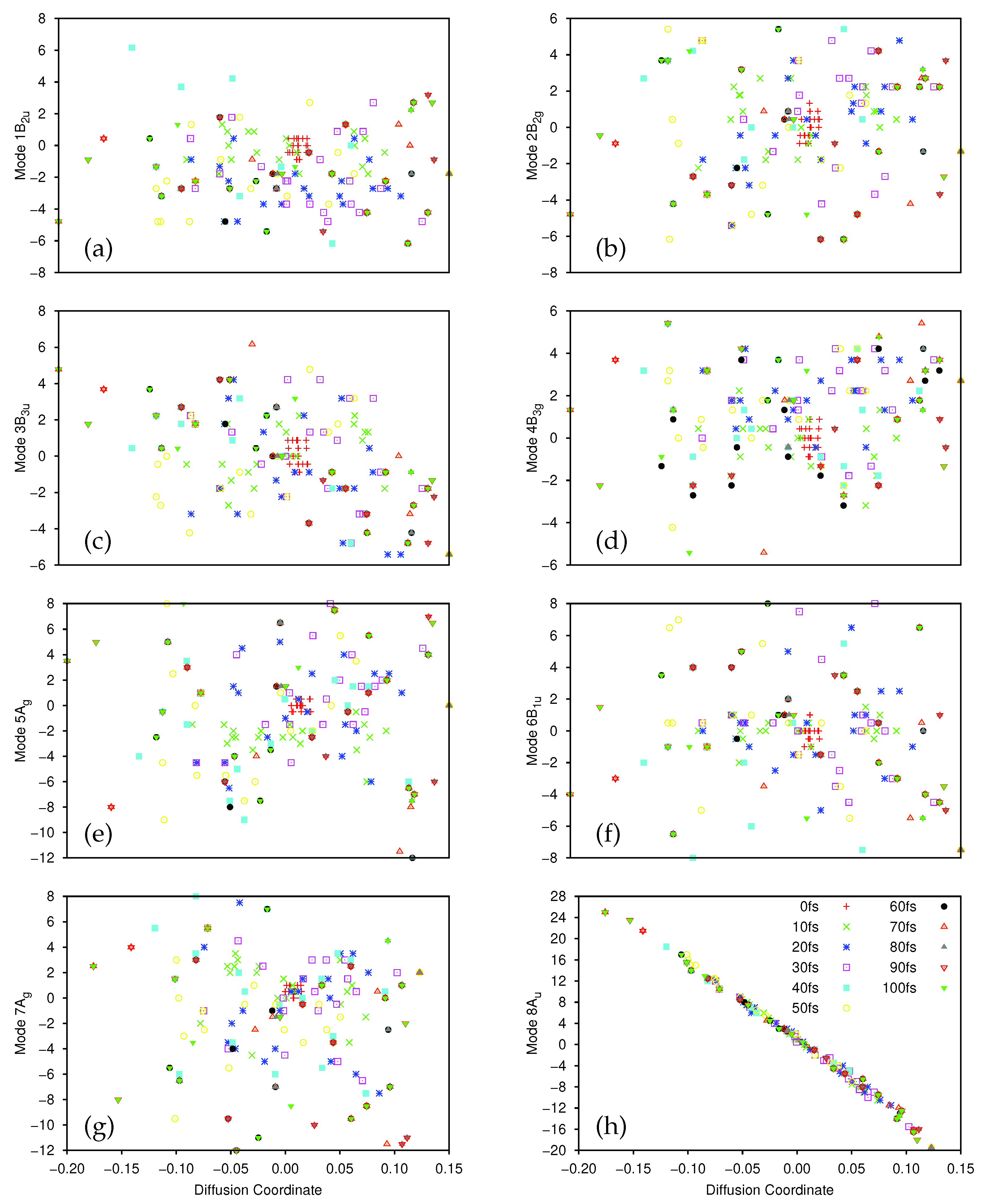
Diffusion map analysis was carried out on the results of the second dynamics calculation for the ground and first exited states using the same parameters as for the first calculation. For the ground state the largest five, non-trivial eigenvalues of the diffusion operator were 0.163, 0.147, 0.136, 0.131 and 0.0926, and for the excited state they were 0.157, 0.137, 0.0932, 0.0897 and 0.0693. In both cases there is no clear spectral gap in the eigenvalues suggesting that the motion of the molecule cannot be reduced to a single mode as was the case for the original calculations; this may be due to the lack of convergence of the dynamics because of the reduction in the density of the gridpoints, such that the gap is not observed. The diffusion map analysis was repeated with lower values of
D in an attempt to locate a spectral gap in line with the results in
Table 3, but none was found, therefore we have to consider at least the five diffusion coordinates with the largest eigenvalues as contributing to the dynamics.
On the excited state, diffusion coordinate correlates most strongly with mode 9B and more weakly with 12B, both of which are C-H stretches, whilst torsion mode 8A correlates with . Coordinate is weakly correlated with the three modes, 8A, 9B and 11B, and correlates with the C-C stretching mode, 7A. The diffusion coordinate with lowest eigenvalue in this selection, , is weakly correlated with modes 2B, 5A and 6B, none of which play a great role in reaching the TPCI, as is the case with and its mode, 7A. However, we see that the three diffusion coordinates with the largest eigenvalues contain contributions from the torsion mode and three of the C-H stretch modes. The absence of 10A is surprising, but may be related to the poor convergence of the dynamics.
On the ground state, is correlated with modes 2B, 4B and 8A, whilst correlates most strongly with the torsion mode and less so with 4B and 11B. C-H stretching mode 12B correlates strongly with coordinate , whilst has contributions from the three C-H stretches excluding 10A, and, finally, is correlated with mode 7A.
From the second calculation on 12D ethene we see from the diffusion map analysis, especially on the excited state, that the torsion mode and three of the C-H stretching modes are important, and these modes also appear in the diffusion coordinates on the ground state; this is indicative of some motion towards the TPCI. The picture is not clear due to the lack of a spectral gap, but we gain useful (if incomplete, with the missing 10A) information about the dynamics towards the conical intersection, even with an unconverged calculation. Longer time propagations, in both the first and second calculations, may resolve more clearly the modes involved.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
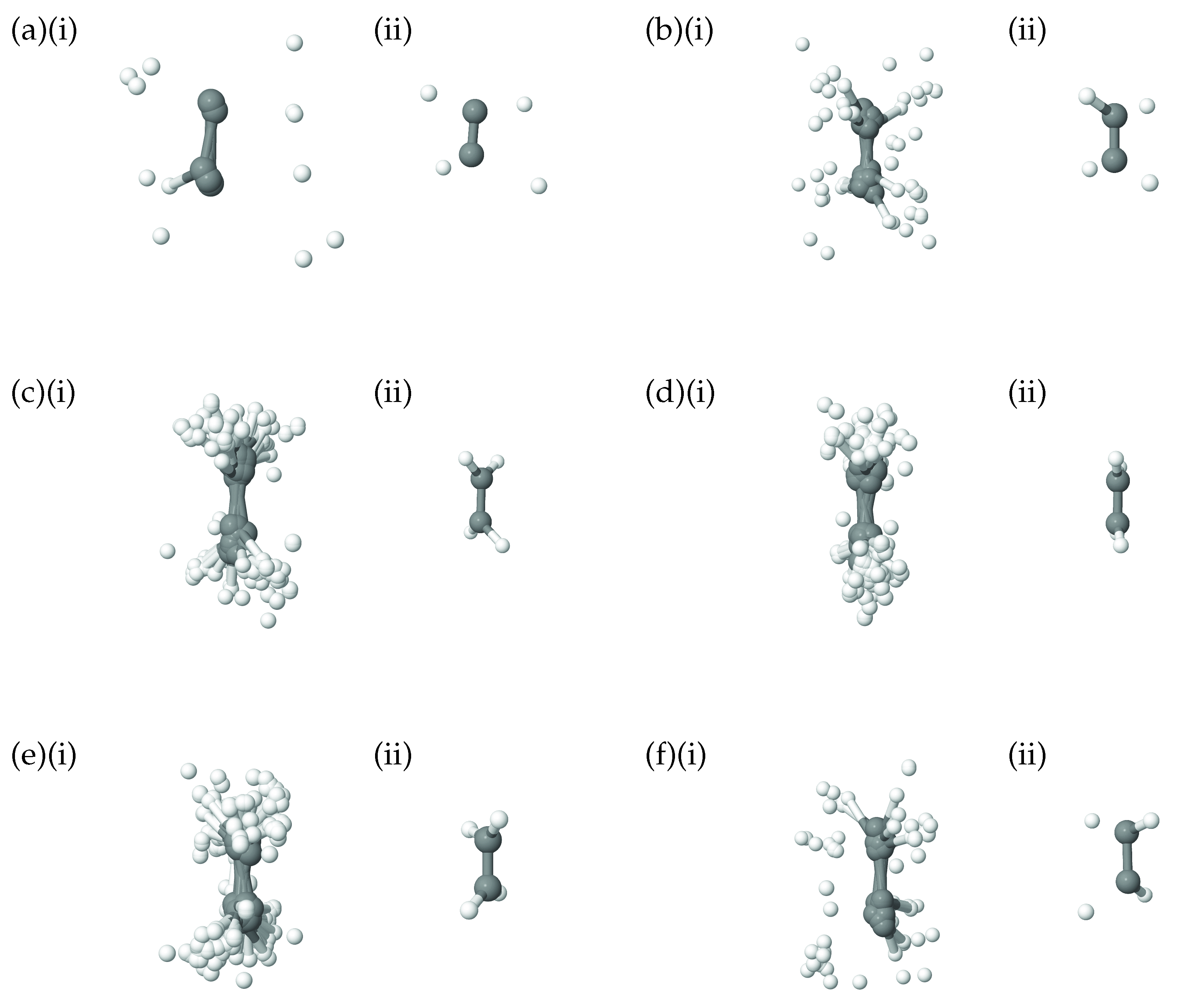{kind=link}
{kind=link}