
DFT-D4 Insight into the Inclusion of Amphetamine and Methamphetamine in Cucurbit[7]uril: Energetic, Structural and Biosensing Properties

 , , and
, , and
Abstract
:1. Introduction
2. Computational Procedures
3. Results and Discussions
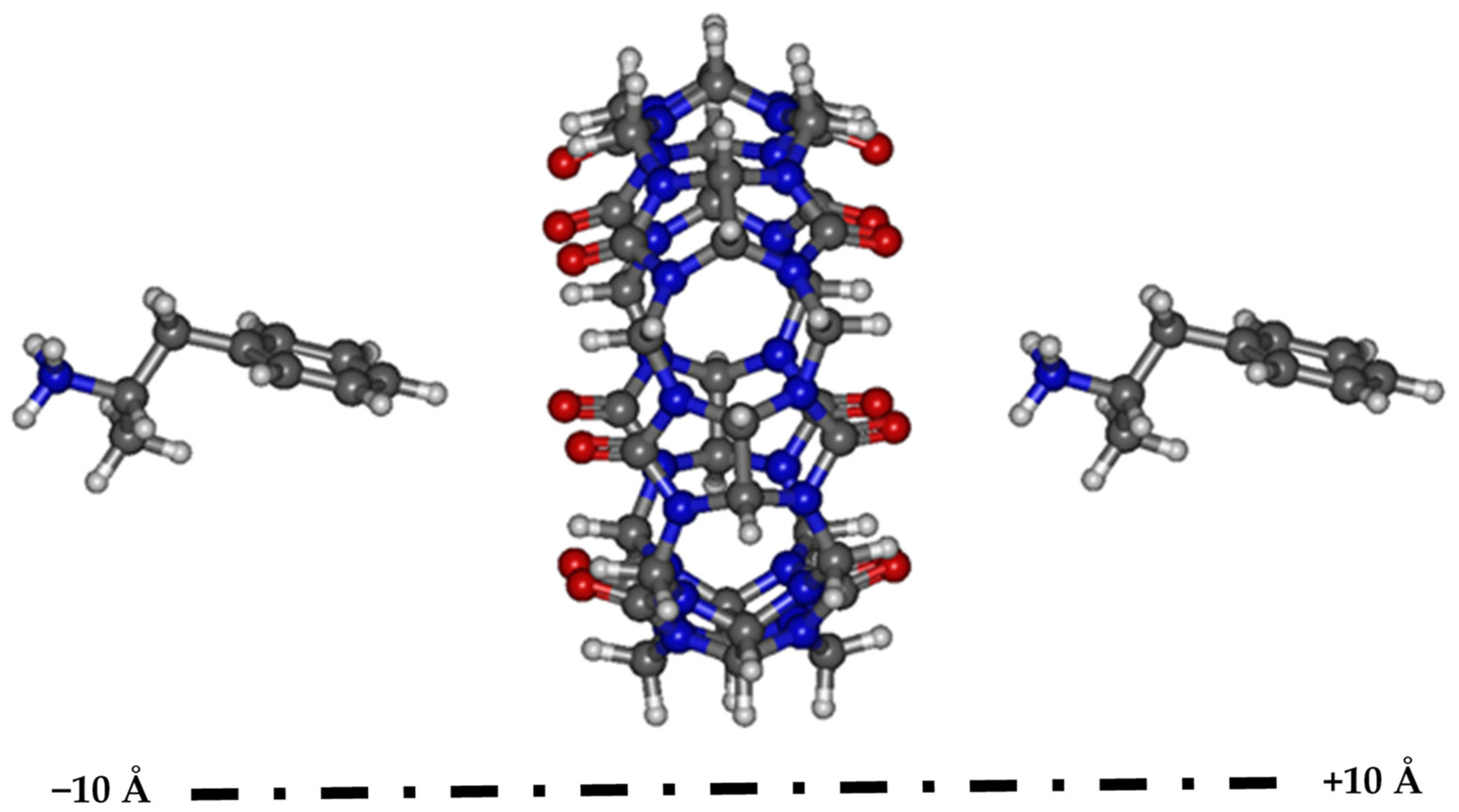
3.1. Searching for the Most Stable Complexes
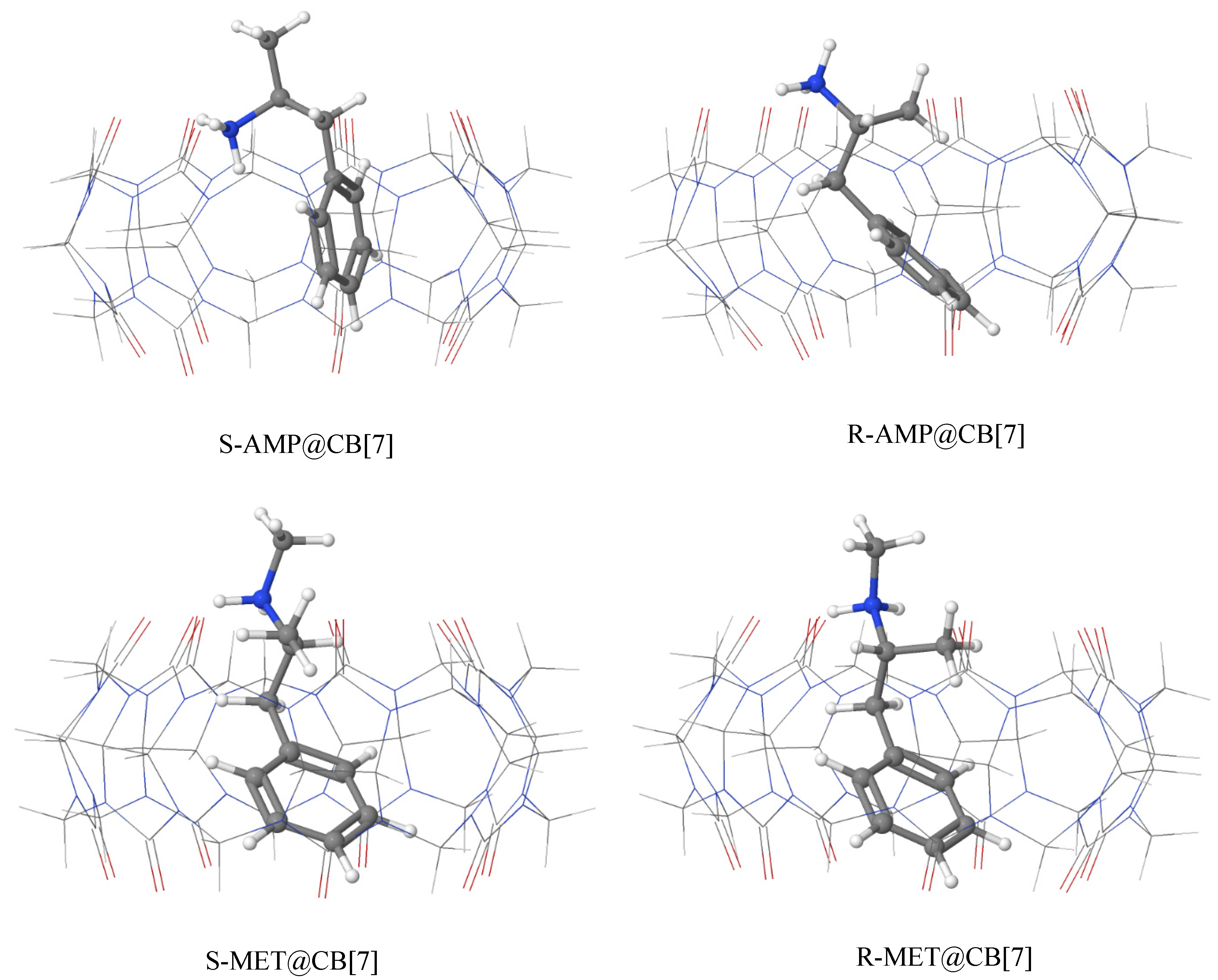
3.2. Geometries of the Most Stable Complexes of AMP and MET with CB[7]
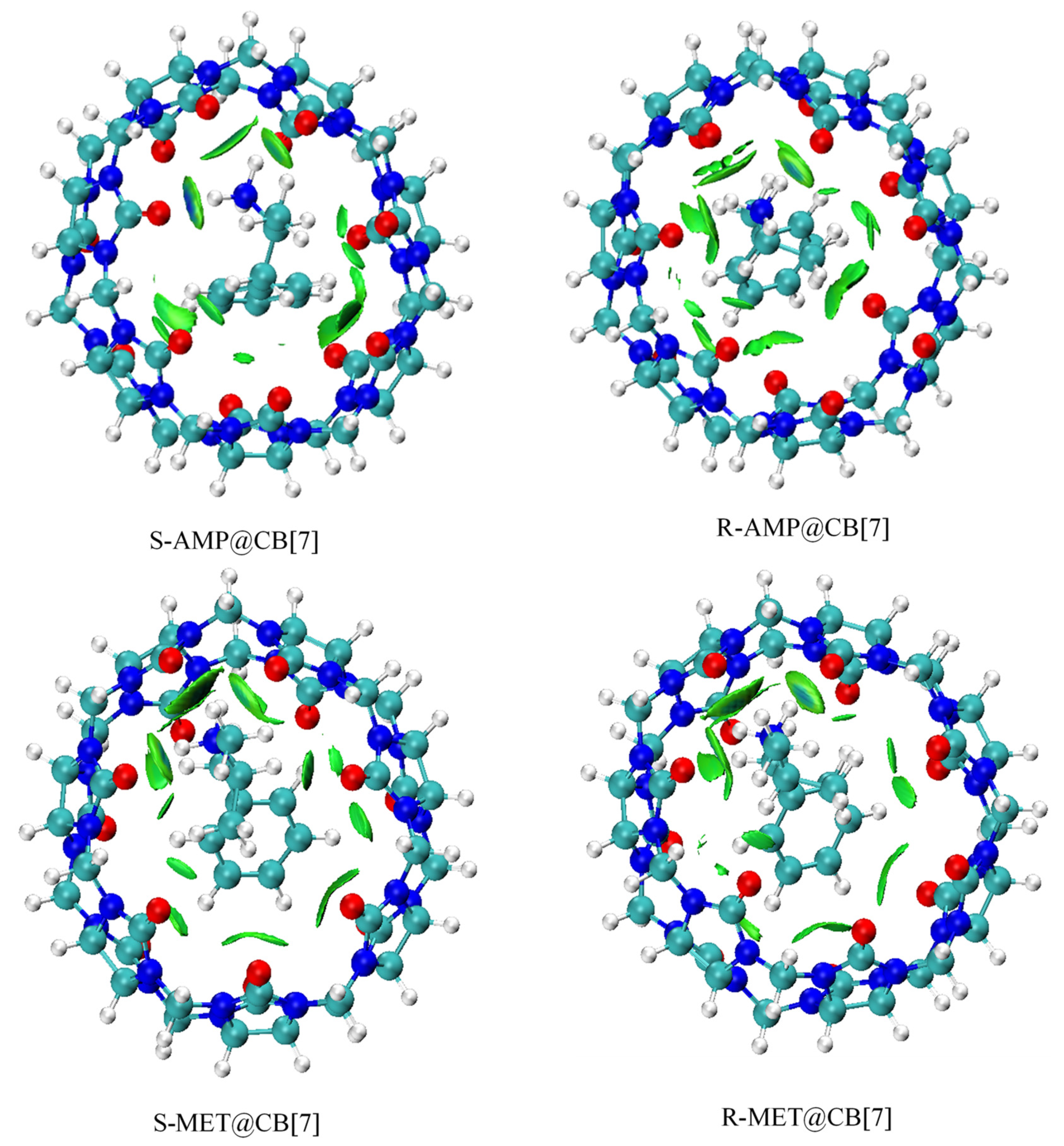
3.3. Analysis of Non-Covalent Interactions
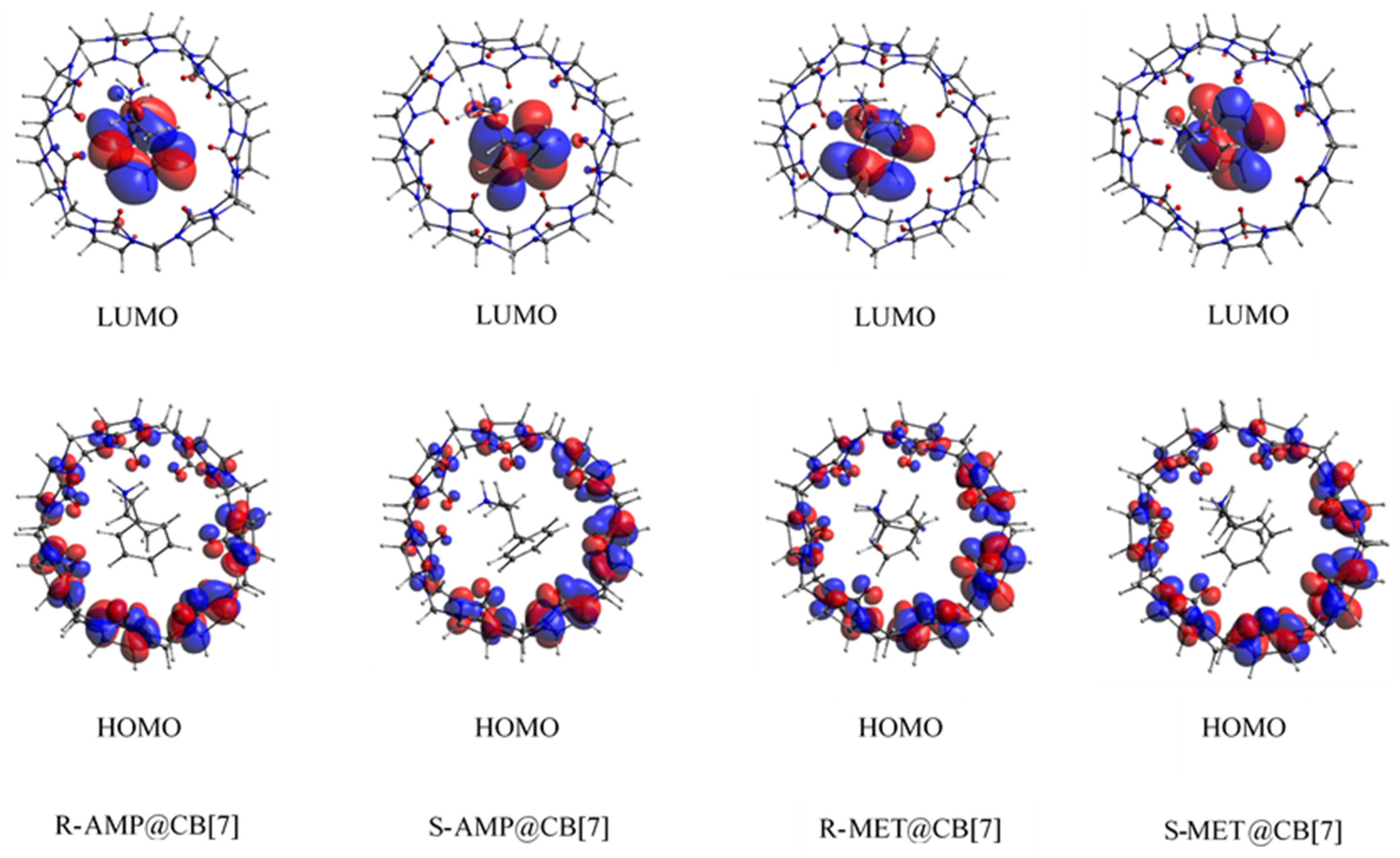
3.4. Biosensing Properties
3.5. Charge Decomposition and Extended Charge Decomposition Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Heal, D.J.; Smith, S.L.; Gosden, J.; Nutt, D.J. Amphetamine, past and present–a pharmacological and clinical perspective. J. Psychopharmacol. 2013, 27, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Sitte, H.H.; Freissmuth, M. Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol. Sci. 2015, 36, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Ruan, X.; Xing, L.; Peng, J.; Li, S.; Song, Y.; Sun, Q. A simplified fabric phase sorptive extraction method for the determination of amphetamine drugs in water samples using liquid chromatography-mass spectrometry. RSC Adv. 2020, 10, 10854–10866. [Google Scholar] [CrossRef]
- Faraone, S.V. The pharmacology of amphetamine and methylphenidate: Relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci. Biobehav. Rev. 2018, 87, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Oei, J.L.; Kingsbury, A.; Dhawan, A.; Burns, L.; Feller, J.M.; Clews, S.; Falconer, J.; Abdel-Latif, M. Amphetamines, the pregnant woman and her children: A review. J. Perinatol. 2012, 32, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodzon-Kulakowska, A.; Antolak, A.; Drabik, A.; Marszalek-Grabska, M.; Kotlińska, J.; Suder, P. Brain lipidomic changes after morphine, cocaine and amphetamine administration—DESI—MS imaging study. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 686–691. [Google Scholar] [CrossRef]
- Berman, S.; O’Neill, J.; Fears, S.; Bartzokis, G.; London, E.D. Abuse of amphetamines and structural abnormalities in brain. Ann. N. Y. Acad. Sci. 2008, 1141, 195. [Google Scholar] [CrossRef] [Green Version]
- Avois, L.; Robinson, N.; Saudan, C.; Baume, N.; Mangin, P.; Saugy, M. Central nervous system stimulants and sport practice. Br. J. Sports Med. 2006, 40, i16–i20. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Management of substance abuse—amphetamine-type stimulants. Available online: https://www.who.int/home/cms-decommissioning (accessed on 30 March 2021).
- Cruickshank, C.C.; Dyer, K.R. A review of the clinical pharmacology of methamphetamine. Addiction 2009, 104, 1085–1099. [Google Scholar] [CrossRef]
- Courtney, K.E.; Ray, L.A. Methamphetamine: An update on epidemiology, pharmacology, clinical phenomenology, and treatment literature. Drug Alcohol Depend. 2014, 143, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Smart, R.G.; Ogborne, A.C. Drug use and drinking among students in 36 countries. Addict. Behav. 2000, 25, 455–460. [Google Scholar] [CrossRef]
- Chomchai, C.; Chomchai, S. Global patterns of methamphetamine use. Curr. Opin. Psychiatry 2015, 28, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Nieddu, M.; Burrai, L.; Baralla, E.; Pasciu, V.; Varoni, M.V.; Briguglio, I.; Demontis, M.P.; Boatto, G. ELISA detection of 30 new amphetamine designer drugs in whole blood, urine and oral fluid using Neogen®“amphetamine” and “methamphetamine/MDMA” kits. J. Anal. Toxicol. 2016, 40, 492–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, L. The validity of self-reported drug use in survey research: An overview and critique of research methods. NIDA Res Monogr 1997, 167, 17–36. [Google Scholar]
- Yu, J.; Qi, D.; Li, J. Design, synthesis and applications of responsive macrocycles. Commun. Chem. 2020, 3, 1–14. [Google Scholar] [CrossRef]
- Sakagami, R.; Saito, Y.; Mori, R.; Satake, M.; Okayasu, M.; Kikkawa, S.; Hikawa, H.; Azumaya, I. Cylindrical macrocyclic compounds synthesized by connecting two bowl-shaped calix[3]aramide moieties: Structures and chiroptical properties. RSC Adv. 2020, 10, 34549–34555. [Google Scholar] [CrossRef]
- Hussain, A.; Yousuf, S.; Mukherjee, D. Importance and synthesis of benzannulated medium-sized and macrocyclic rings (BMRs). RSC Adv. 2014, 4, 43241–43257. [Google Scholar] [CrossRef]
- Jie, K.; Zhou, Y.; Yao, Y.; Huang, F. Macrocyclic amphiphiles. Chem. Soc. Rev. 2015, 44, 3568–3587. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Samal, S.; Selvapalam, N.; Kim, H.-J.; Kim, K. Cucurbituril homologues and derivatives: New opportunities in supramolecular chemistry. Acc. Chem. Res. 2003, 36, 621–630. [Google Scholar] [CrossRef]
- Masson, E.; Ling, X.; Joseph, R.; Kyeremeh-Mensah, L.; Lu, X. Cucurbituril chemistry: A tale of supramolecular success. Rsc Adv. 2012, 2, 1213–1247. [Google Scholar] [CrossRef]
- Kim, J.; Jung, I.-S.; Kim, S.-Y.; Lee, E.; Kang, J.-K.; Sakamoto, S.; Yamaguchi, K.; Kim, K. New cucurbituril homologues: Syntheses, isolation, characterization, and X-ray crystal structures of cucurbit[n]uril (n = 5, 7, and 8). J. Am. Chem. Soc. 2000, 122, 540–541. [Google Scholar] [CrossRef]
- Kircheva, N.; Dobrev, S.; Dasheva, L.; Koleva, I.; Nikolova, V.; Angelova, S.; Dudev, T. Complexation of biologically essential (mono-and divalent) metal cations to cucurbiturils: A DFT/SMD evaluation of the key factors governing the host–guest recognition. RSC Adv. 2020, 10, 28139–28147. [Google Scholar] [CrossRef]
- Liu, S.; Zavalij, P.Y.; Isaacs, L. Cucurbit[10]uril. J. Am. Chem. Soc. 2005, 127, 16798–16799. [Google Scholar] [CrossRef] [PubMed]
- Assaf, K.I.; Nau, W.M. Cucurbiturils: From synthesis to high-affinity binding and catalysis. Chem. Soc. Rev. 2015, 44, 394–418. [Google Scholar] [CrossRef] [Green Version]
- Honda, Y.; Hanaya, T.; Sueishi, Y. Inclusion complexation abilities of cucurbit[6]uril for various aromatic amines in the presence of alkali metal cations. J. Incl. Phenom. Macrocycl. Chem. 2017, 88, 253–257. [Google Scholar] [CrossRef]
- Zhang, S.; Grimm, L.; Miskolczy, Z.; Biczók, L.; Biedermann, F.; Nau, W.M. Binding affinities of cucurbit[n]urils with cations. Chem. Commun. 2019, 55, 14131–14134. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Kim, S.-Y.; Ko, Y.H.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Novel molecular drug carrier: Encapsulation of oxaliplatin in cucurbit[7]uril and its effects on stability and reactivity of the drug. Org. Biomol. Chem. 2005, 3, 2122–2125. [Google Scholar] [CrossRef] [Green Version]
- Saleh, N.I.; Khaleel, A.; Al-Dmour, H.; Al-Hindawi, B.; Yakushenko, E. Host–guest complexes of cucurbit[7]uril with albendazole in solid state. J. Therm. Anal. Calorim. 2013, 111, 385–392. [Google Scholar] [CrossRef]
- Uzunova, V.D.; Cullinane, C.; Brix, K.; Nau, W.M.; Day, A.I. Toxicity of cucurbit[7]uril and cucurbit[8]uril: An exploratory in vitro and in vivo study. Org. Biomol. Chem. 2010, 8, 2037–2042. [Google Scholar] [CrossRef]
- Zhang, P.; Qin, S.; Qi, M.; Fu, R. Cucurbit[n]urils as a new class of stationary phases for gas chromatographic separations. J. Chromatogr. A 2014, 1334, 139–148. [Google Scholar] [CrossRef]
- Jang, Y.; Jang, M.; Kim, H.; Lee, S.J.; Jin, E.; Koo, J.Y.; Hwang, I.-C.; Kim, Y.; Ko, Y.H.; Hwang, I. Point-of-use detection of amphetamine-type stimulants with host-molecule-functionalized organic transistors. Chem 2017, 3, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Hao, H.; Qin, A.; Tang, B.Z. Highly sensitive chemosensor for detection of methamphetamine by the combination of AIE luminogen and cucurbit[7]uril. Dye. Pigment. 2020, 180, 108413. [Google Scholar] [CrossRef]
- Assaba, I.M.; Rahali, S.; Belhocine, Y.; Allal, H. Inclusion complexation of chloroquine with α and β-cyclodextrin: Theoretical insights from the new B97-3c composite method. J. Mol. Struct. 2021, 1227, 129696. [Google Scholar] [CrossRef]
- Allal, H.; Belhocine, Y.; Rahali, S.; Damous, M.; Ammouchi, N. Structural, electronic, and energetic investigations of acrolein adsorption on B 36 borophene nanosheet: A dispersion-corrected DFT insight. J. Mol. Modeling 2020, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bouhadiba, A.; Rahali, S.; Belhocine, Y.; Allal, H.; Nouar, L.; Rahim, M. Structural and energetic investigation on the host/guest inclusion process of benzyl isothiocyanate into β-cyclodextrin using dispersion-corrected DFT calculations. Carbohydr. Res. 2020, 491, 107980. [Google Scholar] [CrossRef] [PubMed]
- Rahali, S.; Belhocine, Y.; Seydou, M.; Maurel, F.; Tangour, B. Multiscale study of the structure and hydrogen storage capacity of an aluminum metal-organic framework. Int. J. Hydrog. Energy 2017, 42, 15271–15282. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Li, F.; Chen, Z. Appropriate description of intermolecular interactions in the methane hydrates: An assessment of DFT methods. J. Comput. Chem. 2013, 34, 121–131. [Google Scholar] [CrossRef]
- Fang, D.; Piquemal, J.-P.; Liu, S.; Cisneros, G.A. DFT-steric-based energy decomposition analysis of intermolecular interactions. Theor. Chem. Acc. 2014, 133, 1484. [Google Scholar] [CrossRef]
- Rahali, S.; Belhocine, Y.; Touzeau, J.; Tangour, B.; Maurel, F.; Seydou, M. Balance between physical and chemical interactions of second-row diatomic molecules with graphene sheet. Superlattices Microstruct. 2017, 102, 45–55. [Google Scholar] [CrossRef]
- Rahali, S.; Seydou, M.; Belhocine, Y.; Maurel, F.; Tangour, B. First-principles investigation of hydrogen storage on lead (II)-based metal-organic framework. Int. J. Hydrogen Energy 2016, 41, 2711–2719. [Google Scholar] [CrossRef]
- Imane, D.; Leila, N.; Fatiha, M.; Abdelkrim, G.; Mouna, C.; Ismahan, L.; Abdelazize, B.; Brahim, H. Investigation of intermolecular interactions in inclusion complexes of pyroquilon with cucurbit[n]urils (n = 7, 8) using DFT-D3 correction dispersion. J. Mol. Liq. 2020, 309, 113233. [Google Scholar] [CrossRef]
- PubChem. Available online: https://www.pubchem.ncbi.nlm.nih.gov/ (accessed on 3 June 2021).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Mewes, J.-M.; Ehlert, S.; Grimme, S. Extension and evaluation of the D4 London-dispersion model for periodic systems. Phys. Chem. Chem. Phys. 2020, 22, 8499–8512. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Kruse, H.; Grimme, S. A geometrical correction for the inter-and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 04B613. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Guo, Q.-X. Use of quantum chemical methods to study cyclodextrin chemistry. J. Incl. Phenom. Macrocycl. Chem. 2004, 50, 95–103. [Google Scholar] [CrossRef]
- Sun, Z.; Huai, Z.; He, Q.; Liu, Z. A General Picture of Cucurbit[8]uril Host–Guest Binding. J. Chem. Inf. Modeling 2021. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Macaya, L.; Vöhringer-Martinez, E. Molecular Environment-Specific Atomic Charges Improve Binding Affinity Predictions of SAMPL5 Host–Guest Systems. J. Chem. Inf. Modeling 2021, 61, 4462–4474. [Google Scholar] [CrossRef]
- Huai, Z.; Yang, H.; Li, X.; Sun, Z. SAMPL7 TrimerTrip host–guest binding affinities from extensive alchemical and end-point free energy calculations. J. Comput.-Aided Mol. Des. 2021, 35, 117–129. [Google Scholar] [CrossRef]
- Yin, J.; Henriksen, N.M.; Slochower, D.R.; Shirts, M.R.; Chiu, M.W.; Mobley, D.L.; Gilson, M.K. Overview of the SAMPL5 Host-Guest Challenge: Are We Doing Better? J. Comput.-Aided Mol. Des. 2017, 31, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzi, A.; Murkli, S.; McNeill, J.N.; Yao, W.; Sullivan, M.; Gilson, M.K.; Chiu, M.W.; Isaacs, L.; Gibb, B.C.; Mobley, D.L.; et al. Overview of the SAMPL6 host– guest binding affinity prediction challenge. J. Comput.-Aided Mol. Des. 2018, 32, 937–963. [Google Scholar] [CrossRef]
- Sun, Z. SAMPL7 TrimerTrip Host-Guest Binding Poses and Binding Affinities from Spherical-Coordinates-Biased Simulations. J. Comput.-Aided Mol. Des. 2021, 35, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Allouche, A.R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Takano, Y.; Houk, K. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The independent gradient model: A new approach for probing strong and weak interactions in molecules from wave function calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Boto, R.A.; Izquierdo-Ruiz, F.; Reva, I.; Woller, T.; Alonso, M. A benchmark for the non-covalent interaction (NCI) index or… is it really all in the geometry? Theor. Chem. Acc. 2016, 135, 242. [Google Scholar] [CrossRef] [Green Version]
- Al-Hamdani, Y.S.; Tkatchenko, A. Understanding non-covalent interactions in larger molecular complexes from first principles. J. Chem. Phys. 2019, 150, 010901. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of donor-acceptor interactions: A charge decomposition analysis using fragment molecular orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Xiao, M.; Lu, T. Generalized charge decomposition analysis (GCDA) method. J. Adv. Phys. Chem 2015, 4, 111–124. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Ghosh, S.; Solomon, E.I. Mechanism of N2O reduction by the μ4-S tetranuclear CuZ cluster of nitrous oxide reductase. J. Am. Chem. Soc. 2006, 128, 278–290. [Google Scholar] [CrossRef]
- Guo, X.; Wang, Z.; Zuo, L.; Zhou, Z.; Guo, X.; Sun, T. Quantitative prediction of enantioseparation using β-cyclodextrin derivatives as chiral selectors in capillary electrophoresis. Analyst 2014, 139, 6511–6519. [Google Scholar] [CrossRef]
- Rezac, J. Non-Covalent Interactions Atlas Benchmark Data Sets 2: Hydrogen Bonding in an Extended Chemical Space. J. Chem. Theory Comput. 2020, 16, 6305–6316. [Google Scholar] [CrossRef] [PubMed]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 6 July 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Configurations | R-AMP@CB[7] | S-AMP@CB[7] | R-MET@CB[7] | S-MET@CB[7] |
|---|---|---|---|---|
| −10 Å | −301.08 | −323.76 | −334.75 | −284.33 |
| −8 Å | −299.69 | −295.34 | −269.57 | −331.66 |
| −6 Å | −324.09 | −346.79 | −335.39 | −320.60 |
| −4 Å | −324.33 | −346.60 | −334.80 | −336.52 |
| −2 Å | −337.34 | −337.14 | −334.73 | −317.27 |
| 0 Å | −337.43 | −337.02 | −334.71 | −317.46 |
| +2 Å | −320.76 | −337.54 | −334.64 | −322.59 |
| +4 Å | −300.51 | −300.90 | −282.05 | −276.98 |
| +6 Å | −300.58 | −300.91 | −277.80 | −291.80 |
| +8 Å | −300.60 | −289.05 | −278.07 | −287.32 |
| +10 Å | −300.71 | −295.21 | −278.39 | −286.65 |
| Complex | ΔEComplexation (kJ/mol) | ΔGComplexation (kJ/mol) (a) | ||
|---|---|---|---|---|
| Gas Phase | Aqueous Phase | Experimental | Calculated | |
| R-AMP@CB[7] | −338.85 | −73.23 | −34.7 | −45.5 |
| S-AMP@CB[7] | −349.58 | −76.41 | ||
| R-MET@CB[7] | −334.01 | −84.01 | −33.8 | −35.3 |
| S-MET@CB[7] | −334.53 | −85.32 | ||
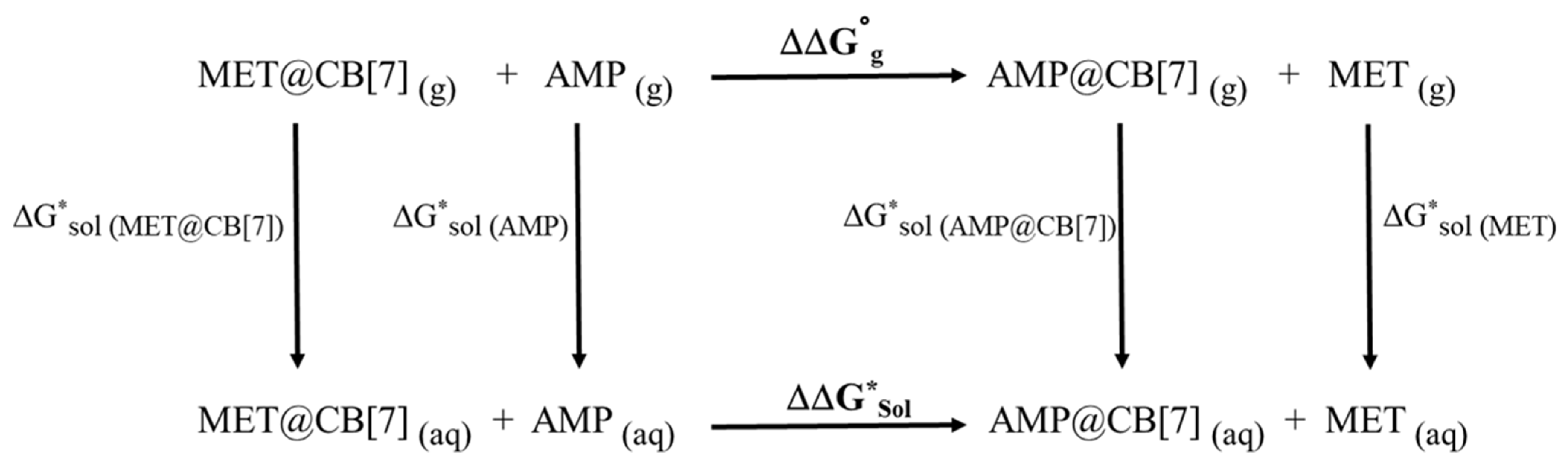
| Species | ΔG0gas (a.u.) | ΔG*sol (a.u.) | ΔΔG0gas (kJ/mol) | ΔΔG*sol (kJ/mol) |
|---|---|---|---|---|
| AMP | −405.16178 | −405.26800 | −10.2 | 12.7 |
| MET | −444.38724 | −444.48477 | ||
| AMP@CB[7] | −4612.20227 | −4612.38314 | ||
| MET@CB[7] | −4651.42384 | −4651.604737 |
| System | EHOMO (eV) | ELUMO (eV) | ∆ELUMO-HOMO (eV) |
|---|---|---|---|
| CB[7] | −5.62 | −0.52 | 5.10 |
| R-AMP@CB[7] | −5.68 | −0.95 | 4.73 |
| S-AMP@CB[7] | −5.68 | −1.13 | 4.55 |
| R-MET@CB[7] | −5.67 | −0.95 | 4.72 |
| S-MET@CB[7] | −5.68 | −0.98 | 4.70 |
| CDA | ECDA | ||||
|---|---|---|---|---|---|
| System | d | b | d-b | r | Net Electrons Obtained by AMP or MET |
| R-AMP@CB[7] | 0.232 | 0.071 | 0.161 | −0.320 | 0.196 |
| S-AMP@CB[7] | 0.209 | 0.042 | 0.167 | −0.306 | 0.206 |
| R-MET@CB[7] | 0.232 | 0.069 | 0.163 | −0.323 | 0.199 |
| S-MET@CB[7] | 0.231 | 0.065 | 0.166 | −0.335 | 0.203 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litim, A.; Belhocine, Y.; Benlecheb, T.; Ghoniem, M.G.; Kabouche, Z.; Ali, F.A.M.; Abdulkhair, B.Y.; Seydou, M.; Rahali, S. DFT-D4 Insight into the Inclusion of Amphetamine and Methamphetamine in Cucurbit[7]uril: Energetic, Structural and Biosensing Properties. Molecules 2021, 26, 7479. https://doi.org/10.3390/molecules26247479
Litim A, Belhocine Y, Benlecheb T, Ghoniem MG, Kabouche Z, Ali FAM, Abdulkhair BY, Seydou M, Rahali S. DFT-D4 Insight into the Inclusion of Amphetamine and Methamphetamine in Cucurbit[7]uril: Energetic, Structural and Biosensing Properties. Molecules. 2021; 26(24):7479. https://doi.org/10.3390/molecules26247479
Chicago/Turabian StyleLitim, Abdelkarim, Youghourta Belhocine, Tahar Benlecheb, Monira Galal Ghoniem, Zoubir Kabouche, Fatima Adam Mohamed Ali, Babiker Yagoub Abdulkhair, Mahamadou Seydou, and Seyfeddine Rahali. 2021. "DFT-D4 Insight into the Inclusion of Amphetamine and Methamphetamine in Cucurbit[7]uril: Energetic, Structural and Biosensing Properties" Molecules 26, no. 24: 7479. https://doi.org/10.3390/molecules26247479
APA StyleLitim, A., Belhocine, Y., Benlecheb, T., Ghoniem, M. G., Kabouche, Z., Ali, F. A. M., Abdulkhair, B. Y., Seydou, M., & Rahali, S. (2021). DFT-D4 Insight into the Inclusion of Amphetamine and Methamphetamine in Cucurbit[7]uril: Energetic, Structural and Biosensing Properties. Molecules, 26(24), 7479. https://doi.org/10.3390/molecules26247479