
Genome-Guided Discovery of the First Myxobacterial Biarylitide Myxarylin Reveals Distinct C–N Biaryl Crosslinking in RiPP Biosynthesis

 ,
,  ,
,
Abstract
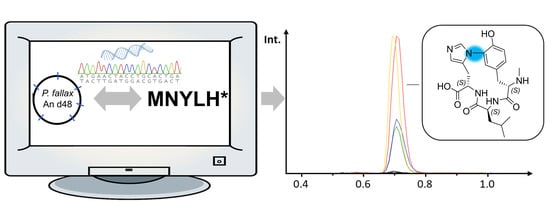
:
1. Introduction
2. Results and Discussion
2.1. Discovery and Purification of the Myxobacterial Biarylitide MeYLH
2.2. Structure Elucidation of 1 and 2
2.3. Heterologous Expression and Biosynthesis
3. Materials and Methods
3.1. Applied Software, DNA Sequence Analysis and Bioinformatic Methods
3.2. Myxobacterial Fermentation and Extraction Procedure for LC-MS Analysis
3.3. Standardized UHPLC-MS Conditions
3.4. Isolation of 1 by Supernatant Derivatization and Semi-Preparative HPLC
3.5. Structure Elucidation
3.5.1. NMR Conditions and Spectroscopic Data
3.5.2. Elucidation of the Absolute Stereochemistry
3.6. Assessment of Antimicrobial Activities
3.7. Molecular Cloning, Construction of Plasmids and Maintenance of Bacterial Cultures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cao, P.; Dey, A.; Vassallo, C.N.; Wall, D. How Myxobacteria Cooperate. J. Mol. Biol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Dorado, J.; Marcos-Torres, F.J.; García-Bravo, E.; Moraleda-Muñoz, A.; Pérez, J. Myxobacteria: Moving, killing, feeding, and surviving together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, J.; Fayad, A.A.; Müller, R. Natural products from myxobacteria: Novel metabolites and bioactivities. Nat. Prod. Rep. 2017, 34, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Müller, R. Myxobacteria—‘Microbial factories’ for the production of bioactive secondary metabolites. Mol. Biosyst. 2009, 5, 567–574. [Google Scholar] [CrossRef]
- Panter, F.; Bader, C.D.; Müller, R. Synergizing the potential of bacterial genomics and metabolomics to find novel antibiotics. Chem. Sci. 2021, 5994–6010. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- De Jong, A.; van Hijum, S.A.F.T.; Bijlsma, J.J.E.; Kok, J.; Kuipers, O.P. BAGEL: A web-based bacteriocin genome mining tool. Nucleic Acids Res. 2006, 34, W273–W279. [Google Scholar] [CrossRef]
- Agrawal, P.; Khater, S.; Gupta, M.; Sain, N.; Mohanty, D. RiPPMiner: A bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res. 2017, 45, W80–W88. [Google Scholar] [CrossRef] [Green Version]
- Santos-Aberturas, J.; Chandra, G.; Frattaruolo, L.; Lacret, R.; Pham, T.H.; Vior, N.M.; Eyles, T.H.; Truman, A.W. Uncovering the unexplored diversity of thioamidated ribosomal peptides in Actinobacteria using the RiPPER genome mining tool. Nucleic Acids Res. 2019, 47, 4624–4637. [Google Scholar] [CrossRef]
- Kloosterman, A.M.; Shelton, K.E.; van Wezel, G.P.; Medema, M.H.; Mitchell, D.A. RRE-Finder: A Genome-Mining Tool for Class-Independent RiPP Discovery. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Merwin, N.J.; Mousa, W.K.; Dejong, C.A.; Skinnider, M.A.; Cannon, M.J.; Li, H.; Dial, K.; Gunabalasingam, M.; Johnston, C.; Magarvey, N.A. DeepRiPP integrates multiomics data to automate discovery of novel ribosomally synthesized natural products. Proc. Natl. Acad. Sci. USA 2020, 117, 371–380. [Google Scholar] [CrossRef]
- De Los Santos, E.L.C. NeuRiPP: Neural network identification of RiPP precursor peptides. Sci. Rep. 2019, 9, 13406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietz, J.I.; Schwalen, C.J.; Patel, P.S.; Maxson, T.; Blair, P.M.; Tai, H.-C.; Zakai, U.I.; Mitchell, D.A. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol. 2017, 13, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.H.; Truman, A.W. Genome mining strategies for ribosomally synthesised and post-translationally modified peptides. Comput. Struct. Biotechnol. J. 2020. [Google Scholar] [CrossRef]
- Hirsch, H.J. Bacteriocins from Myxococcus fulvus (Myxobacterales). Arch. Microbiol. 1977, 115, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.; Hirsch, H.-J. The primary structure of fulvocin C from Myxococcus fulvus. Biochim. Biophys. Acta Protein Struct. 1981, 667, 213–217. [Google Scholar] [CrossRef]
- Mccurdy, H.D.; MacRae, T.H. Xanthacin. A bacteriocin of Myxococcus xanthus fb. Can. J. Microbiol. 1974, 20, 131–135. [Google Scholar] [CrossRef]
- Viehrig, K.; Surup, F.; Volz, C.; Herrmann, J.; Abou Fayad, A.; Adam, S.; Kohnke, J.; Trauner, D.; Müller, R. Structure and biosynthesis of crocagins: Polycyclic postranslationally modified ribosomal peptides from Chondromyces crocatus. Angew. Chem. 2017, 1–5. [Google Scholar] [CrossRef]
- Adam, S.; Klein, A.; Surup, F.; Koehnke, J. The structure of CgnJ, a domain of unknown function protein from the crocagin gene cluster. Acta Crystallogr. F Struct. Biol. Commun. 2019, 75, 205–211. [Google Scholar] [CrossRef]
- Hug, J.J.; Dastbaz, J.; Adam, S.; Revermann, O.; Koehnke, J.; Krug, D.; Müller, R. Biosynthesis of Cittilins, Unusual Ribosomally Synthesized and Post-translationally Modified Peptides from Myxococcus xanthus. ACS Chem. Biol. 2020, 15, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Trowitzsch-Kienast, W. (Ed.) Cittilins: Bicyclic Isotrityrosines from Myxococcus xanthus. In Proceedings of the 24th General Meeting German Chemists’ Society, Hamburg, Germany, 5–11 September 1993. [Google Scholar]
- Reichenbach, H.; Höfle, G. Myxobacteria as producers of secondary metabolites. In Drug Discovery from Nature; Grabley, S., Thiericke, R., Eds.; Springer: Berlin, Germany, 1999; pp. 149–179. [Google Scholar]
- Zdouc, M.M.; Alanjary, M.M.; Zarazúa, G.S.; Maffioli, S.I.; Crüsemann, M.; Medema, M.H.; Donadio, S.; Sosio, M. A biaryl-linked tripeptide from Planomonospora reveals a widespread class of minimal RiPP gene clusters. Cell Chem. Biol. 2020. [Google Scholar] [CrossRef]
- Chai, Y.; Pistorius, D.; Ullrich, A.; Weissman, K.J.; Kazmaier, U.; Müller, R. Discovery of 23 natural tubulysins from Angiococcus disciformis An d48 and Cystobacter SBCb004. Chem. Biol. 2010, 17, 296–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panter, F.; Krug, D.; Baumann, S.; Müller, R. Self-resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria. Chem. Sci. 2018, 9, 4898–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandmann, A.; Frank, B.; Müller, R. A transposon-based strategy to scale up myxothiazol production in myxobacterial cell factories. J. Biotechnol. 2008, 135, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.M.; Bradshaw, E.; Seipke, R.F.; Hutchings, M.I.; McArthur, M. Use and discovery of chemical elicitors that stimulate biosynthetic gene clusters in Streptomyces bacteria. Methods Enzymol. 2012, 517, 367–385. [Google Scholar] [CrossRef]
- Seyedsayamdost, M.R. High-throughput platform for the discovery of elicitors of silent bacterial gene clusters. Proc. Natl. Acad. Sci. USA 2014, 111, 7266–7271. [Google Scholar] [CrossRef] [Green Version]
- De Felício, R.; Ballone, P.; Bazzano, C.F.; Alves, L.F.G.; Sigrist, R.; Infante, G.P.; Niero, H.; Rodrigues-Costa, F.; Fernandes, A.Z.N.; Tonon, L.A.C.; et al. Chemical Elicitors Induce Rare Bioactive Secondary Metabolites in Deep-Sea Bacteria under Laboratory Conditions. Metabolites 2021, 11, 107. [Google Scholar] [CrossRef]
- Hug, J.J.; Müller, R. Host Development for Heterologous Expression and Biosynthetic Studies of Myxobacterial Natural Products: 6.09. In Comprehensive Natural Products III; Liu, H.-W.B., Begley, T.P., Eds.; Elsevier: Oxford, UK, 2020; pp. 149–216. ISBN 978-0-08-102691-5. [Google Scholar]
- B’Hymer, C.; Montes-Bayon, M.; Caruso, J.A. Marfey’s reagent: Past, present, and future uses of 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide. J. Sep. Sci. 2003, 26, 7–19. [Google Scholar] [CrossRef]
- Pogorevc, D.; Müller, R. Biotechnological production optimization of argyrins—A potent immunomodulatory natural product class. Microb. Biotechnol. 2021. [Google Scholar] [CrossRef]
- Phan, C.-S.; Matsuda, K.; Balloo, N.; Fujita, K.; Wakimoto, T.; Okino, T. Argicyclamides A-C Unveil Enzymatic Basis for Guanidine Bis-prenylation. J. Am. Chem. Soc. 2021, 143, 10083–10087. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Linington, R.G. npatlas—The Natural Products Atlas. Available online: https://www.npatlas.org (accessed on 22 November 2021).
- Van Santen, J.A.; Jacob, G.; Singh, A.L.; Aniebok, V.; Balunas, M.J.; Bunsko, D.; Neto, F.C.; Castaño-Espriu, L.; Chang, C.; Clark, T.N.; et al. The natural products atlas: An open access knowledge base for microbial natural products discovery. ACS Cent. Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Kashefi, K.; Hartzell, P.L. Genetic suppression and phenotypic masking of a Myxococcus xanthus frzF-defect. Mol. Microbiol. 1995, 15, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Hug, J.J.; Panter, F.; Krug, D.; Müller, R. Genome mining reveals uncommon alkylpyrones as type III PKS products from myxobacteria. J. Ind. Microbiol. Biotechnol. 2019, 46, 319–334. [Google Scholar] [CrossRef]
- Pogorevc, D.; Panter, F.; Schillinger, C.; Jansen, R.; Wenzel, S.C.; Müller, R. Production optimization and biosynthesis revision of corallopyronin A, a potent anti-filarial antibiotic. Metab. Eng. 2019, 55, 201–211. [Google Scholar] [CrossRef]
- Magrini, V.; Creighton, C.; Youderian, P. Site-specific recombination of temperate Myxococcus xanthus phage Mx8: genetic elements required for integration. J. Bacteriol. 1999, 181, 4050–4061. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganism | MIC of 1 | MIC of 2 |
|---|---|---|
| Acinetobacter baumanii DSM 30008 | >64 | >64 |
| Mucor hiemalis DSM 2656 | >64 | >64 |
| Cryptococcus neoformans DSM 11959 | >64 | >64 |
| Staphylococcus aureus Newman | >64 | >64 |
| Pseudomonas aeruginosa PA14 (DSM 19882) | >64 | >64 |
| Escherichia coli acrB JW0451-2 | >64 | >64 |
| E. coli wild-type BW25113 (DSM 27469) | >64 | >64 |
| Bacillus subtilis DSM 10 | >64 | >64 |
| Candida albicans DSM 1665 | >64 | >64 |
| Pichia anomala DSM 6766 | >64 | >64 |
| Citrobacter freundii DSM 30039 | >64 | >64 |
| Mycobacterium smegmatis MC2 155 | >64 | >64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hug, J.J.; Frank, N.A.; Walt, C.; Šenica, P.; Panter, F.; Müller, R. Genome-Guided Discovery of the First Myxobacterial Biarylitide Myxarylin Reveals Distinct C–N Biaryl Crosslinking in RiPP Biosynthesis. Molecules 2021, 26, 7483. https://doi.org/10.3390/molecules26247483
Hug JJ, Frank NA, Walt C, Šenica P, Panter F, Müller R. Genome-Guided Discovery of the First Myxobacterial Biarylitide Myxarylin Reveals Distinct C–N Biaryl Crosslinking in RiPP Biosynthesis. Molecules. 2021; 26(24):7483. https://doi.org/10.3390/molecules26247483
Chicago/Turabian StyleHug, Joachim J., Nicolas A. Frank, Christine Walt, Petra Šenica, Fabian Panter, and Rolf Müller. 2021. "Genome-Guided Discovery of the First Myxobacterial Biarylitide Myxarylin Reveals Distinct C–N Biaryl Crosslinking in RiPP Biosynthesis" Molecules 26, no. 24: 7483. https://doi.org/10.3390/molecules26247483
APA StyleHug, J. J., Frank, N. A., Walt, C., Šenica, P., Panter, F., & Müller, R. (2021). Genome-Guided Discovery of the First Myxobacterial Biarylitide Myxarylin Reveals Distinct C–N Biaryl Crosslinking in RiPP Biosynthesis. Molecules, 26(24), 7483. https://doi.org/10.3390/molecules26247483