
Decoding the Structure of Non-Proteinogenic Amino Acids: The Rotational Spectrum of Jet-Cooled Laser-Ablated Thioproline

 ,
,  ,
, 

Abstract
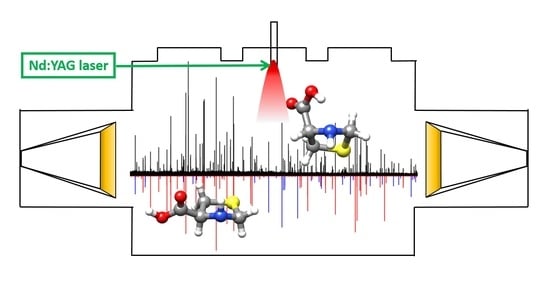
:
1. Introduction
2. Results and Discussion
2.1. Conformational Panorama
2.2. Rotational Spectra
2.3. Conformer Assignment
2.4. Intramolecular Interactions
2.5. Comparison with Related Systems
3. Methods
3.1. Experimental
3.2. Theoretical
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Young, T.S.; Schultz, P.G. Beyond the canonical 20 amino acids: Expanding the genetic lexicon. J. Biol. Chem. 2010, 285, 11039–11044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salwiczek, M.; Nyakatura, E.K.; Gerling, U.I.M.; Ye, S.; Koksch, B. Fluorinated amino acids: Compatibility with native protein structures and effects on protein–protein interactions. Chem. Soc. Rev. 2012, 41, 2135–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.K.; Naduthambi, D.; Thomas, K.M.; Zondlo, N.J. Proline editing: A general and practical approach to the synthesis of functionally and structurally diverse peptides. Analysis of steric versus stereoelectronic effects of 4-substituted prolines on conformation within peptides. J. Am. Chem. Soc. 2013, 135, 4333–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner-White, E.J.; Bell, L.H.; Maccallum, P.H. Pyrrolidine ring puckering in cis and trans-proline residues in proteins and polypeptides. Different puckers are favoured in certain situations. J. Mol. Biol. 1992, 228, 725–734. [Google Scholar] [CrossRef]
- Kim, M.K.; Kang, Y.K. Positional preference of proline in α-helices. Protein Sci. 1999, 8, 1492–1499. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Satyshur, K.A.; Forest, K.T.; Raines, R.T. Stereoelectronic and steric effects in side chains preorganize a protein main chain. Proc. Natl. Acad. Sci. USA 2010, 107, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Lesarri, A.; Cocinero, E.J.; López, J.C.; Alonso, J.L. Shape of 4(S)- and 4(R)-hydroxyproline in gas phase. J. Am. Chem. Soc. 2005, 127, 2572–2579. [Google Scholar] [CrossRef]
- Bretscher, L.E.; Jenkins, C.L.; Taylor, K.M.; DeRider, M.L.; Raines, R.T. Conformational stability of collagen relies on a stereoelectronic effect. J. Am. Chem. Soc. 2001, 123, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.L.; Raines, R.T. Insights on the conformational stability of collagen. Nat. Prod. Rep. 2002, 19, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Rather, S.; Clarke, H.T. The Action of Formaldehyde upon Cysteine. J. Am. Chem. Soc. 1937, 59, 200–206. [Google Scholar] [CrossRef]
- Weber, H.U.; Fleming, J.F.; Miquel, J. Thiazolidine-4-carboxylic acid, a physiologic sulfhydryl antioxidant with potential value in geriatric medicine. Arch. Gerontol. Geriatr. 1982, 1, 299–310. [Google Scholar] [CrossRef]
- De La Fuente, M.; Ferrández, M.D.; Del Rio, M.; Sol Burgos, M.; Miquel, J. Enhancement of leukocyte functions in aged mice supplemented with the antioxidant thioproline. Mech. Ageing Dev. 1998, 104, 213–225. [Google Scholar] [CrossRef]
- Roberts, J.C.; Nagasawa, H.T.; Zera, R.T.; Fricke, R.F.; Goon, D.J.W. Prodrugs of L-Cysteine as Protective Agents against Acetaminophen-Induced Hepatotoxicity: 2-(Polyhydroxyalkyl)- and 2-(Polyacetoxyalkyl)thiazolidine-4(R)-carboxylic Acids. J. Med. Chem. 1987, 30, 1891–1896. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.; Del Río, M.; De La Fuente, M. Improvement of murine immune functions in vitro by thioproline. Immunopharmacology 1999, 44, 281–291. [Google Scholar] [CrossRef]
- Chao, T.F.; Leu, H.B.; Huang, C.C.; Chen, J.W.; Chan, W.L.; Lin, S.J.; Chen, S.A. Thiazolidinediones can prevent new onset atrial fibrillation in patients with non-insulin dependent diabetes. Int. J. Cardiol. 2012, 156, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Ramírez-Boscá, A.; Ramírez-Bosca, J.V.; Alperi, J.D. Menopause: A review on the role of oxygen stress and favorable effects of dietary antioxidants. Arch. Gerontol. Geriatr. 2006, 42, 289–306. [Google Scholar] [CrossRef]
- Frank, N.; Tsuda, M.; Ohgaki, H.; Frei, E.; Kato, T.; Sato, S. Detoxifying potential of thioproline against N-nitroso compounds, N-nitrosodimethylamine and N-nitrosocimetidine. Cancer Lett. 1990, 50, 167–172. [Google Scholar] [CrossRef]
- Kurashima, Y.; Tsuda, M.; Sugimura, T. Marked Formation of Thiazolidine-4-carboxylic Acid, an Effective Nitrite Trapping Agent in Vivo, on Boiling of Dried Shiitake Mushroom (Lentinus edodes). J. Agric. Food Chem. 1990, 38, 1945–1949. [Google Scholar] [CrossRef]
- Suvachittanont, W.; Kurashima, Y.; Esumi, H.; Tsuda, M. Formation of thiazolidine-4-carboxylic acid (thioproline), an effective nitrite-trapping agent in human body, in Parkia speciosa seeds and other edible leguminous seeds in Thailand. Food Chem. 1996, 55, 359–363. [Google Scholar] [CrossRef]
- Liu, J.; Chan, W. Quantification of thiazolidine-4-carboxylic acid in toxicant-exposed cells by isotope-dilution liquid chromatography-mass spectrometry reveals an intrinsic antagonistic response to oxidative stress-induced toxicity. Chem. Res. Toxicol. 2015, 28, 394–400. [Google Scholar] [CrossRef]
- Liu, J.; Chan, K.K.J.; Chan, W. Identification of Protein Thiazolidination as a Novel Molecular Signature for Oxidative Stress and Formaldehyde Exposure. Chem. Res. Toxicol. 2016, 29, 1865–1871. [Google Scholar] [CrossRef]
- Ham, Y.H.; Jason Chan, K.K.; Chan, W. Thioproline Serves as an Efficient Antioxidant Protecting Human Cells from Oxidative Stress and Improves Cell Viability. Chem. Res. Toxicol. 2020, 33, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hao, C.; Wu, L.; Chan, W.; Lam, H. Proteomic analysis of thioproline misincorporation in Escherichia coli. J. Proteom. 2020, 210, 103541. [Google Scholar] [CrossRef]
- Baldwin, E.T.; Bhat, T.N.; Gulnik, S.; Liu, B.; Topol, I.A.; Kiso, Y.; Mimoto, T.; Mitsuya, H.; Erickson, J.W. Structure of HIV-1 protease with KNI-272, a tight-binding transition-state analog containing allophenylnorstatine. Structure 1995, 3, 581–590. [Google Scholar] [CrossRef]
- Doi, M.; Ishida, T.; Katsuya, Y.; Sasaki, M.; Taniguchi, T.; Hasegawa, H.; Mimoto, T.; Kiso, Y. KNI-272, a highly selective and potent peptidic HIV protease inhibitor. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2001, 57, 1333–1335. [Google Scholar] [CrossRef]
- Murcko, M.A.; Rao, B.G.; Gomperts, R. Conformational analysis of HIV-1 protease inhibitors: 2. Thioproline P1? Residue in the potent inhibitor KNI-272. J. Comput. Chem. 1997, 18, 1151–1166. [Google Scholar] [CrossRef]
- David, L.; Luo, R.; Head, M.S.; Gilson, M.K. Computational study of KNI-272, a potent inhibitor of HIV-1 protease: On the mechanism of preorganization. J. Phys. Chem. B 1999, 103, 1031–1044. [Google Scholar] [CrossRef]
- Adachi, M.; Ohhara, T.; Kurihara, K.; Tamada, T.; Honjo, E.; Okazaki, N.; Arai, S.; Shoyama, Y.; Kimura, K.; Matsumura, H.; et al. Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proc. Natl. Acad. Sci. USA 2009, 106, 4641–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kánai, K.; Podányi, B.; Bokotey, S.; Hajdú, F.; Hermecz, I. Stereoselective sulfoxide formation from a thioproline derivative. Tetrahedron Asymmetry 2002, 13, 491–495. [Google Scholar] [CrossRef]
- Grant, N.; Ward, M.F.; Jaspars, M.; Harrison, W.T.A. (R)-1,3-Thiazolidin-3-ium-4-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2001, 57, o697–o699. [Google Scholar] [CrossRef] [Green Version]
- Lesarri, A.; Mata, S.; Cocinero, E.J.; Blanco, S.; López, J.C.; Alonso, J.L. The structure of neutral proline. Angew. Chem. Int. Ed. 2002, 41, 4673–4676. [Google Scholar] [CrossRef]
- Mata, S.; Vaquero, V.; Cabezas, C.; Pena, I.; Perez, C.; Lopez, J.C.; Alonso, J.L. Observation of two new conformers of neutral proline. Phys. Chem. Chem. Phys. 2009, 11, 4141–4144. [Google Scholar] [CrossRef] [PubMed]
- Grabow, J.-U.; Caminati, W. Microwave Spectroscopy: Experimental Techniques. Front. Mol. Spectrosc. 2009, 2009, 383–454. [Google Scholar] [CrossRef]
- Pate, B.H. Taking the Pulse of Molecular Rotational Spectroscopy. Science 2011, 333, 947–948. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Talebi, M.; Thakur, N.; Wahab, M.F.; Mikhonin, A.V.; Muckle, M.T.; Neill, J.L. A Gas Chromatography-Molecular Rotational Resonance Spectroscopy Based System of Singular Specificity. Angew. Chem. Int. Ed. 2020, 59, 192–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugachandran, S.I.; Tang, J.; Peña, I.; Loru, D.; Sanz, M.E. New Insights into Secondary Organic Aerosol Formation: Water Binding to Limonene. J. Phys. Chem. Lett. 2021, 12, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.L.; López, J.C. Microwave spectroscopy of biomolecular building blocks. Top. Curr. Chem. 2015, 364, 335–401. [Google Scholar] [CrossRef]
- Blanco, S.; Macario, A.; López, J.C. The structure of isolated thalidomide as reference for its chirality-dependent biological activity: A laser-ablation rotational study. Phys. Chem. Chem. Phys. 2021, 23, 13705–13713. [Google Scholar] [CrossRef] [PubMed]
- Caminati, W.; Di Bernardo, S. Microwave spectrum and ring puckering motion in thiazolidine. J. Mol. Spectrosc. 1989, 137, 354–361. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Pickett, H.M. The fitting and prediction of vibration-rotation spectra with spin interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure a Series of Advances; Durig, J.R., Ed.; Elsevier: New York, NY, USA, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Gordy, W.; Cook, R.L. Microwave Molecular Spectra; Wiley-Interscience: New York, NY, USA, 1984; Volume 11, ISBN 0471086819. [Google Scholar]
- Blanco, S.; Lesarri, A.; López, J.C.; Alonso, J.L. The gas-phase structure of alanine. J. Am. Chem. Soc. 2004, 126, 11675–11683. [Google Scholar] [CrossRef]
- Ruoff, R.S.; Klots, T.D.; Emilsson, T.; Gutowsky, H.S. Relaxation of conformers and isomers in seeded supersonic jets of inert gases. J. Chem. Phys. 1990, 93, 3142–3150. [Google Scholar] [CrossRef]
- Florio, G.M.; Christie, R.A.; Jordan, K.D.; Zwier, T.S. Conformational preferences of jet-cooled melatonin: Probing trans- and cis-amide regions of the potential energy surface. J. Am. Chem. Soc. 2002, 124, 10236–10247. [Google Scholar] [CrossRef]
- Godfrey, P.D.; Brown, R.D. Proportions of species observed in jet spectroscopy-vibrational-energy effects: Histamine tautomers and conformers. J. Am. Chem. Soc. 1998, 120, 10724–10732. [Google Scholar] [CrossRef]
- Blanco, S.; Sanz, M.E.; López, J.C.; Alonso, J.L. Revealing the multiple structures of serine. Proc. Natl. Acad. Sci. USA 2007, 104, 20183–20188. [Google Scholar] [CrossRef] [Green Version]
- Sanz, M.E.; Blanco, S.; Löpez, J.C.; Alonso, J.L. Rotational probes of six conformers of neutral cysteine. Angew. Chem. Int. Ed. 2008, 47, 6216–6220. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piriou, F.; Lintner, K.; Lam-Thanh, H.; Toma, F.; Fermandjian, S. Synthesis of 13c-labelled [s]-proline and its conformational analysis by nuclear magnetic resonance. Tetrahedron 1978, 34, 553–556. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. IUCr The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Pinacho, P.; Blanco, S.; López, J.C. The complete conformational panorama of formanilide–water complexes: The role of water as a conformational switch. Phys. Chem. Chem. Phys. 2019, 21, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Medcraft, C.; Gougoula, E.; Bittner, D.M.; Mullaney, J.C.; Blanco, S.; Tew, D.P.; Walker, N.R.; Legon, A.C. Molecular geometries and other properties of H2O⋯AgI and H3 N⋯AgI as characterised by rotational spectroscopy and ab initio calculations. J. Chem. Phys. 2017, 147, 234308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rotamer a | II-ax-ta | Rotamer b | I-ax-ce | III-ax-ce | |
|---|---|---|---|---|---|
| Param. a | exp | MP2 | exp | MP2 | MP2 |
| A/MHz | 3215.82492(41) b | 3076.88 | 3803.26722(85) | 3806.75 | 3835.54 |
| B/MHz | 1317.61215(14) | 1365.99 | 1091.61856(34) | 1084.75 | 1071.96 |
| C/MHz | 1089.68026(14) | 1136.06 | 957.57523(39) | 960.86 | 980.62 |
| κ | −0.78 | −0.76 | −0.91 | −0.91 | −0.94 |
| DJ/kHz | 1.4116(24) | 0.828 | 0.145(12) | 0.085 | 0.080 |
| DJK/kHz | −9.9744(69) | −5.017 | 0.151(20) | 0.193 | 0.306 |
| DK/kHz | 23.112(71) | 10.195 | [0.0] c | 0.788 | 0.509 |
| d1/kHz | −0.02597(59) | −0.021 | −0.0225(18) | −0.020 | −0.008 |
| d2/kHz | −0.00317(22) | −0.001 | −0.00230(10) | −0.008 | −0.002 |
| 3/2(χaa)/MHz | −1.9796(31) | −2.14 | 3.0136(53) | 2.93 | 2.67 |
| 1/4(χbb-χcc)/MHz | −0.59941(88) | −0.61 | 0.1996(18) | 0.17 | 0.01 |
| χaa/MHz | −1.3193(21) | −1.43 | 2.0091(35) | 1.95 | 1.78 |
| χbb/MHz | −0.5391(28) | −0.51 | −0.6053(54) | −0.63 | −0.87 |
| χcc/MHz | 1.8585(28) | 1.93 | −1.4037(54) | −1.32 | −0.91 |
| n | 199 | 93 | |||
| σ/kHz | 4.2 | 4.9 | |||
| μa,μb,μc/D | 2.3, 2.6, 2.9 | 1.0, 0.7, 0.6 | 0.3, 0.9, 1.3 | ||
| ΔE/cm−1 | 118.4 | 0.0 | 332.5 | ||
| ΔG/cm−1 | 314.5 | 0.0 | 370.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López, J.C.; Macario, A.; Verde, A.; Pérez-Encabo, A.; Blanco, S. Decoding the Structure of Non-Proteinogenic Amino Acids: The Rotational Spectrum of Jet-Cooled Laser-Ablated Thioproline. Molecules 2021, 26, 7585. https://doi.org/10.3390/molecules26247585
López JC, Macario A, Verde A, Pérez-Encabo A, Blanco S. Decoding the Structure of Non-Proteinogenic Amino Acids: The Rotational Spectrum of Jet-Cooled Laser-Ablated Thioproline. Molecules. 2021; 26(24):7585. https://doi.org/10.3390/molecules26247585
Chicago/Turabian StyleLópez, Juan Carlos, Alberto Macario, Andrés Verde, Alfonso Pérez-Encabo, and Susana Blanco. 2021. "Decoding the Structure of Non-Proteinogenic Amino Acids: The Rotational Spectrum of Jet-Cooled Laser-Ablated Thioproline" Molecules 26, no. 24: 7585. https://doi.org/10.3390/molecules26247585
APA StyleLópez, J. C., Macario, A., Verde, A., Pérez-Encabo, A., & Blanco, S. (2021). Decoding the Structure of Non-Proteinogenic Amino Acids: The Rotational Spectrum of Jet-Cooled Laser-Ablated Thioproline. Molecules, 26(24), 7585. https://doi.org/10.3390/molecules26247585