
Applications of Solution NMR in Drug Discovery



Abstract
:1. Introduction
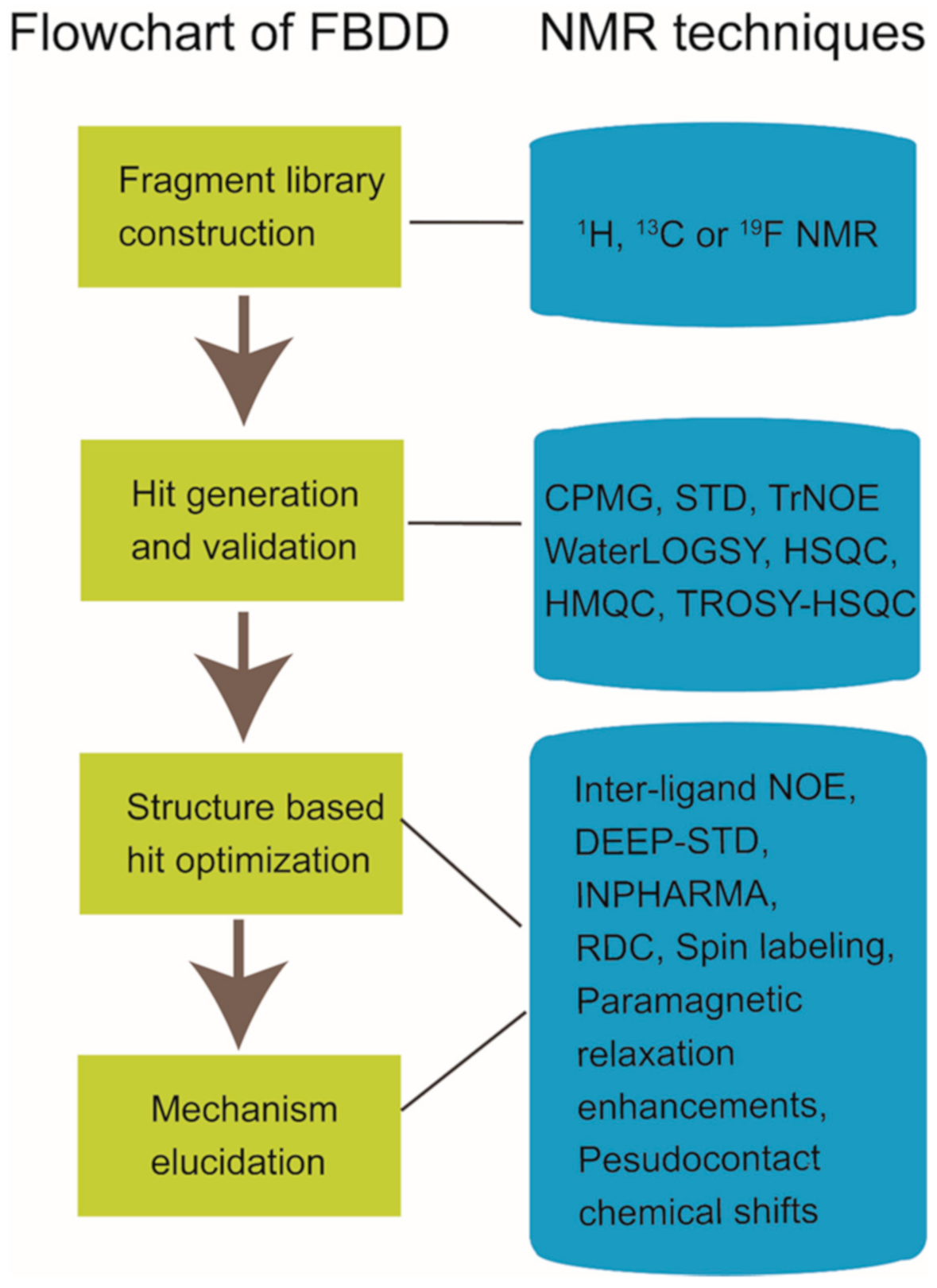
2. NMR in Fragment-Based Drug Discovery
2.1. Fragment Compound Libray Construction and Group Generation
2.2. Ligand-Observed or Target-Observed Hit Generation and Validation
2.3. Hit-to-Lead Optimization
2.4. Working Mechanism Elucidation
3. NMR in PPI Modulators Discovery
4. In-Cell NMR
5. Conclusion Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Emwas, A.H.; Szczepski, K.; Poulson, B.G.; Chandra, K.; McKay, R.T.; Dhahri, M.; Alahmari, F.; Jaremko, L.; Lachowicz, J.I.; Jaremko, M. NMR as a “Gold Standard” Method in Drug Design and Discovery. Molecules 2020, 25, 4597. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kang, C.B. A Practical Perspective on the Roles of Solution NMR Spectroscopy in Drug Discovery. Molecules 2020, 25, 2974. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Bertini, I.; Cowburn, D.; Dalvit, C.; Giralt, E.; Jahnke, W.; James, T.L.; Homans, S.W.; Kessler, H.; Luchinat, C.; et al. Perspectives on NMR in drug discovery: A technique comes of age. Nat. Rev. Drug Discov. 2008, 7, 738–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017, 61, 453–464. [Google Scholar]
- Harner, M.J.; Frank, A.O.; Fesik, S.W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Li, Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Front. Mol. Biosci. 2020, 7, 180. [Google Scholar] [CrossRef]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef]
- Ayotte, Y.; Murugesan, J.R.; Bilodeau, F.; Larda, S.; Bouchard, P.; Drouin, N.; Morin, M.; LaPlante, S. Discovering Quality Drug Seeds by Practical NMR-based Fragment Screening. Protein Sci. 2017, 26, 194–195. [Google Scholar]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Ma, R.; Wang, P.; Wu, J.; Ruan, K. Process of Fragment-Based Lead Discovery-A Perspective from NMR. Molecules 2016, 21, 854. [Google Scholar]
- Yanamala, N.; Dutta, A.; Beck, B.; van Fleet, B.; Hay, K.; Yazbak, A.; Ishima, R.; Doemling, A.; Klein-Seetharaman, J. NMR-based screening of membrane protein ligands. Chem. Biol. Drug Des. 2010, 75, 237–256. [Google Scholar] [PubMed]
- Iconaru, L.I.; Ban, D.; Bharatham, K.; Ramanathan, A.; Zhang, W.; Shelat, A.A.; Zuo, J.; Kriwacki, R.W. Discovery of Small Molecules that Inhibit the Disordered Protein, p27(Kip1). Sci. Rep. 2015, 5, 15686. [Google Scholar] [PubMed] [Green Version]
- Han, B.; Ahn, H.C. Recombinant Kinase Production and Fragment Screening by NMR Spectroscopy. Methods Mol. Biol. 2016, 1360, 35–46. [Google Scholar] [PubMed]
- Binas, O.; de Jesus, V.; Landgraf, T.; Volklein, A.E.; Martins, J.; Hymon, D.; Bains, J.K.; Berg, H.; Biedenbander, T.; Furtig, B.; et al. F-19 NMR-Based Fragment Screening for 14 Different Biologically Active RNAs and 10 DNA and Protein Counter-Screens. ChemBioChem 2020, 22, 423–433. [Google Scholar]
- Keseru, G.M.; Erlanson, D.A.; Ferenczy, G.G.; Hann, M.M.; Murray, C.W.; Pickett, S.D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [PubMed] [Green Version]
- Kobayashi, M.; Retra, K.; Figaroa, F.; Hollander, J.G.; Ab, E.; Heetebrij, R.J.; Irth, H.; Siegal, G. Target Immobilization as a Strategy for NMR-Based Fragment Screening: Comparison of TINS, STD, and SPR for Fragment Hit Identification. J. Biomol. Screen. 2010, 15, 978–989. [Google Scholar]
- Huth, J.R.; Sun, C. Utility of NMR in lead optimization: Fragment-based approaches. Comb. Chem. High Throughput Screen. 2002, 5, 631–643. [Google Scholar]
- Recht, M.I.; Sridhar, V.; Badger, J.; Bounaud, P.Y.; Logan, C.; Chie-Leon, B.; Nienaber, V.; Torres, F.E. Identification and Optimization of PDE10A Inhibitors Using Fragment-Based Screening by Nanocalorimetry and X-ray Crystallography. J. Biomol. Screen. 2014, 19, 497–507. [Google Scholar]
- Stark, J.L.; Powers, R. Application of NMR and molecular docking in structure-based drug discovery. Top. Curr. Chem. 2012, 326, 1–34. [Google Scholar]
- Orts, J.; Gossert, A.D. Structure determination of protein-ligand complexes by NMR in solution. Methods 2018, 138, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Sugiki, T.; Furuita, K.; Fujiwara, T.; Kojima, C. Current NMR Techniques for Structure-Based Drug Discovery. Molecules 2018, 23, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernini, A.; Henrici De Angelis, L.; Morandi, E.; Spiga, O.; Santucci, A.; Assfalg, M.; Molinari, H.; Pillozzi, S.; Arcangeli, A.; Niccolai, N. Searching for protein binding sites from Molecular Dynamics simulations and paramagnetic fragment-based NMR studies. Biochim. Biophys. Acta 2014, 1844, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Softley, C.A.; Bostock, M.J.; Popowicz, G.M.; Sattler, M. Paramagnetic NMR in drug discovery. J. Biomol. NMR 2020, 74, 287–309. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Davis, B.J.; Jahnke, W. Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem. Biol. 2019, 26, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, D.M.; Van Molle, I.; Baud, M.G.; Galdeano, C.; Geraldes, C.F.; Ciulli, A. Is NMR Fragment Screening Fine-Tuned to Assess Druggability of Protein-Protein Interactions? ACS Med. Chem. Lett. 2014, 5, 23–28. [Google Scholar] [CrossRef]
- Valkov, E.; Sharpe, T.; Marsh, M.; Greive, S.; Hyvonen, M. Targeting protein-protein interactions and fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 145–179. [Google Scholar]
- Bosch, J. PPI inhibitor and stabilizer development in human diseases. Drug Discov. Today Technol. 2017, 24, 3–9. [Google Scholar] [CrossRef]
- Lu, H.Y.; Zhou, Q.D.; He, J.; Jiang, Z.L.; Peng, C.; Tong, R.S.; Shi, J.Y. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Li, L.; Jiang, J.; Zheng, Z.; Shang, J.; Wang, C.; Chen, W.; Bao, Q.; Xu, X.; et al. Small-molecule inhibitor targeting the Hsp90-Cdc37 protein-protein interaction in colorectal cancer. Sci. Adv. 2019, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Lu, S.Y.; Zhang, J. Emerging roles of allosteric modulators in the regulation of protein-protein interactions (PPIs): A new paradigm for PPI drug discovery. Med. Res. Rev. 2019, 39, 2314–2342. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, D.; Di Maro, S.; Cerofolini, L.; Giuntini, S.; Fragai, M.; Luchinat, C.; Tomassi, S.; Limatola, A.; Russomanno, P.; Merlino, F.; et al. HOPPI-NMR: Hot-Peptide-Based Screening Assay for Inhibitors of Protein-Protein Interactions by NMR. ACS Med. Chem. Lett. 2020, 11, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Sijbesma, E.; Hallenbeck, K.K.; Leysen, S.; de Vink, P.J.; Skora, L.; Jahnke, W.; Brunsyeld, L.; Arkin, M.R.; Ottmann, C. Site-Directed Fragment-Based Screening for the Discovery of Protein-Protein Interaction Stabilizers. J. Am. Chem. Soc. 2019, 141, 3524–3531. [Google Scholar] [CrossRef]
- Sheng, C.Q.; Dong, G.Q.; Miao, Z.Y.; Zhang, W.N.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors (vol 44, pg 8238, 2015). Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Chen, Y.; Lian, F.; Chen, L.; Li, Y.; Cao, D.; Wang, X.; Chen, L.; Li, J.; Meng, T.; et al. Fragment-based drug discovery of triazole inhibitors to block PDEdelta-RAS protein-protein interaction. Eur. J. Med Chem. 2019, 163, 597–609. [Google Scholar] [CrossRef]
- Kategaya, L.; Di Lello, P.; Rouge, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.P.; Prakash, S.; et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017, 550, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Ceccarelli, D.F.; Orlicky, S.; St-Cyr, D.J.; Ziemba, A.; Garg, P.; Plamondon, S.; Auer, M.; Sidhu, S.; Marinier, A.; et al. E2 enzyme inhibition by stabilization of a low-affinity interface with ubiquitin. Nat. Chem. Biol. 2014, 10, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Milroy, L.G.; Bartel, M.; Henen, M.A.; Leysen, S.; Adriaans, J.M.; Brunsveld, L.; Landrieu, I.; Ottmann, C. Stabilizer-Guided Inhibition of Protein-Protein Interactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 15720–15724. [Google Scholar] [CrossRef]
- Bower, J.F.; Pannifer, A. Using fragment-based technologies to target protein-protein interactions. Curr. Pharm. Des. 2012, 18, 4685–4696. [Google Scholar] [CrossRef] [PubMed]
- Moriya, J.; Takeuchi, K.; Tai, K.J.; Arai, K.; Kobayashi, N.; Yoneda, N.; Fukunishi, Y.; Inoue, A.; Kihara, M.; Murakami, T.; et al. Structure-Based Development of a Protein-Protein Interaction Inhibitor Targeting Tumor Necrosis Factor Receptor-Associated Factor 6. J. Med. Chem. 2015, 58, 5674–5683. [Google Scholar] [CrossRef] [PubMed]
- Kang, C. Applications of In-Cell NMR in Structural Biology and Drug Discovery. Int. J. Mol. Sci. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegal, G.; Selenko, P. Cells, drugs and NMR. J. Magn. Reson. 2019, 306, 202–212. [Google Scholar] [CrossRef]
- Cerofolini, L.; Giuntini, S.; Barbieri, L.; Pennestri, M.; Codina, A.; Fragai, M.; Banci, L.; Luchinat, E.; Ravera, E. Real-Time Insights into Biological Events: In-Cell Processes and Protein-Ligand Interactions. Biophys. J. 2019, 116, 239–247. [Google Scholar] [CrossRef]
- Maldonado, A.Y.; Burz, D.S.; Shekhtman, A. In-cell NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 197–212. [Google Scholar] [CrossRef]
- Luchinat, E.; Banci, L. In-Cell NMR in Human Cells: Direct Protein Expression Allows Structural Studies of Protein Folding and Maturation. Acc. Chem. Res. 2018, 51, 1550–1557. [Google Scholar]
- Selenko, P.; Wagner, G. Looking into live cells with in-cell NMR spectroscopy. J. Struct. Biol. 2007, 158, 244–253. [Google Scholar]
- Li, C.; Zhao, J.; Cheng, K.; Ge, Y.; Wu, Q.; Ye, Y.; Xu, G.; Zhang, Z.; Zheng, W.; Zhang, X.; et al. Magnetic Resonance Spectroscopy as a Tool for Assessing Macromolecular Structure and Function in Living Cells. Annu. Rev. Anal. Chem. 2017, 10, 157–182. [Google Scholar]
- Mashalidis, E.H.; Sledz, P.; Lang, S.; Abell, C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat. Protoc. 2013, 8, 2309–2324. [Google Scholar]
- Osborne, J.; Panova, S.; Rapti, M.; Urushima, T.; Jhoti, H. Fragments: Where are we now? Biochem. Soc. Trans. 2020, 48, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, A.F.; Barrios, A.G. Pushing the Ligand Efficiency Metrics: Relative Group Contribution (RGC) Model as a Helpful Strategy to Promote a Fragment “Rescue” Effect. Front. Chem. 2019, 7, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bembenek, S.D.; Tounge, B.A.; Reynolds, C.H. Ligand efficiency and fragment-based drug discovery. Drug Discov. Today 2009, 14, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, X.; Wang, A.; Zheng, Y.; Gao, Y.; Zhou, J. Evolutions in fragment-based drug design: The deconstruction-reconstruction approach. Drug Discov. Today 2015, 20, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.J.; Mortenson, P.N.; Murray, C.W. Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 2014, 116, 82–91. [Google Scholar] [CrossRef]
- Tanaka, D.; Tsuda, Y.; Shiyama, T.; Nishimura, T.; Chiyo, N.; Tominaga, Y.; Sawada, N.; Mimoto, T.; Kusunose, N. A Practical Use of Ligand Efficiency Indices Out of the Fragment-Based Approach: Ligand Efficiency-Guided Lead Identification of Soluble Epoxide Hydrolase Inhibitors. J. Med. Chem. 2011, 54, 851–857. [Google Scholar] [CrossRef]
- Doak, B.C.; Morton, C.J.; Simpson, J.S.; Scanlon, M.J. Design and Evaluation of the Performance of an NMR Screening Fragment Library. Aust. J. Chem. 2013, 66, 1465–1472. [Google Scholar] [CrossRef]
- Shi, Y.; von Itzstein, M. How Size Matters: Diversity for Fragment Library Design. Molecules 2019, 24, 2838. [Google Scholar] [CrossRef] [Green Version]
- Troelsen, N.S.; Shanina, E.; Gonzalez-Romero, D.; Dankova, D.; Jensen, I.S.A.; Sniady, K.J.; Nami, F.; Zhang, H.; Rademacher, C.; Cuenda, A.; et al. The 3F Library: Fluorinated Fsp(3) -Rich Fragments for Expeditious (19) F NMR Based Screening. Angew. Chem. Int. Ed. Engl. 2020, 59, 2204–2210. [Google Scholar] [CrossRef]
- Liu, M.; Quinn, R.J. Fragment-based screening with natural products for novel anti-parasitic disease drug discovery. Expert Opin. On Drug Discov. 2019, 14, 1283–1295. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B. Broad coverage of commercially available lead-like screening space with fewer than 350,000 compounds. J. Chem. Inf. Modeling 2013, 53, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, M.; Barozet, A.; Johansson, C.; Eriksson, P.O.; Engkvist, O.; Walsh, J.; Nissink, J.W.M. Utility of Resazurin, Horseradish Peroxidase, and NMR Assays to Identify Redox-Related False-Positive Behavior in High-Throughput Screens. Assay Drug Dev. Technol. 2018, 16, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Cuellar, M.; Singh, G.; Nelson, K.M.; Strasser, J.; Rappe, T.; Xia, Y.; Veglia, G.; Walters, M.A. ALARM NMR for HTS triage and chemical probe validation. Curr. Protoc. Chem. Biol. 2018, 10, 91–117. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivoli, M.; Novak, H.R.; Littlechild, J.A.; Harmer, N.J. Determination of protein-ligand interactions using differential scanning fluorimetry. J. Vis. Exp. 2014, 51809. [Google Scholar] [CrossRef] [Green Version]
- Campos-Olivas, R. NMR screening and hit validation in fragment based drug discovery. Curr. Top. Med. Chem. 2011, 11, 43–67. [Google Scholar] [CrossRef]
- Hennig, M.; Ruf, A.; Huber, W. Combining biophysical screening and X-ray crystallography for fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 115–143. [Google Scholar]
- McMahon, R.M.; Scanlon, M.J.; Martin, J.L. Interrogating Fragments Using a Protein Thermal Shift Assay. Aust. J. Chem. 2013, 66, 1502–1506. [Google Scholar] [CrossRef] [Green Version]
- Linke, P.; Amaning, K.; Maschberger, M.; Vallee, F.; Steier, V.; Baaske, P.; Duhr, S.; Breitsprecher, D.; Rak, A. An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J. Biomol. Screen. 2016, 21, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.L.; Chen, T.T.; Zhou, C.; Lian, F.L.; Tang, X.L.; Wen, Y.; Shen, J.K.; Xu, Y.C.; Xiong, B.; Zhang, N.X. NMR-based platform for fragment-based lead discovery used in screening BRD4-targeted compounds. Acta Pharmacol. Sin. 2016, 37, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, X.; Goldflam, M.; Feliz, M.; Belda, I.; Giralt, E. Computer-aided design of fragment mixtures for NMR-based screening. PLoS ONE 2013, 8, e58571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, J.L.; Eghbalnia, H.R.; Lee, W.; Westler, W.M.; Markley, J.L. NMRmix: A Tool for the Optimization of Compound Mixtures in 1D (1)H NMR Ligand Affinity Screens. J. Proteome Res. 2016, 15, 1360–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, R.S.; Leung, E.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of (19)F-NMR in Fragment-Based Drug Discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Vulpetti, A. Ligand-Based Fluorine NMR Screening: Principles and Applications in Drug Discovery Projects. J. Med. Chem. 2019, 62, 2218–2244. [Google Scholar] [CrossRef] [PubMed]
- Andersen, N.; Clausen, M. Synthesis and 19F NMR-based screening of a library of diverse and three-dimensional fluorinated fragments. Abstr. Pap. Am. Chem. Soc. 2018, 256. [Google Scholar]
- Jordan, J.B.; Poppe, L.; Xia, X.; Cheng, A.C.; Sun, Y.; Michelsen, K.; Eastwood, H.; Schnier, P.D.; Nixey, T.; Zhong, W. Fragment based drug discovery: Practical implementation based on F-19 NMR spectroscopy. J. Med. Chem. 2012, 55, 678–687. [Google Scholar] [CrossRef]
- Dalvit, C.; Vulpetti, A. Technical and practical aspects of (19) F NMR-based screening: Toward sensitive high-throughput screening with rapid deconvolution. Magn. Reson. Chem. 2012, 50, 592–597. [Google Scholar] [CrossRef]
- Sreeramulu, S.; Richter, C.; Kuehn, T.; Azzaoui, K.; Blommers, M.J.J.; Del Conte, R.; Fragai, M.; Trieloff, N.; Schmieder, P.; Nazare, M.; et al. NMR quality control of fragment libraries for screening. J. Biomol. NMR 2020, 74, 555–563. [Google Scholar] [CrossRef]
- Neumann, T.; Junker, H.D.; Schmidt, K.; Sekul, R. SPR-based fragment screening: Advantages and applications. Curr. Top. Med. Chem. 2007, 7, 1630–1642. [Google Scholar] [CrossRef]
- Maple, H.J.; Garlish, R.A.; Rigau-Roca, L.; Porter, J.; Whitcombe, I.; Prosser, C.E.; Kennedy, J.; Henry, A.J.; Taylor, R.J.; Crump, M.P.; et al. Automated protein-ligand interaction screening by mass spectrometry. J. Med. Chem. 2012, 55, 837–851. [Google Scholar] [CrossRef]
- Duong-Thi, M.D.; Meiby, E.; Bergstrom, M.; Fex, T.; Isaksson, R.; Ohlson, S. Weak affinity chromatography as a new approach for fragment screening in drug discovery. Anal. Biochem. 2011, 414, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, S.; Duong-Thi, M.D. Fragment screening for drug leads by weak affinity chromatography (WAC-MS). Methods 2018, 146, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Olson, N.M.; Tooker, M.J.; Bur, S.K.; Pomerantz, W.C.K. Combined Protein- and Ligand-Observed NMR Workflow to Screen Fragment Cocktails against Multiple Proteins: A Case Study Using Bromodomains. Molecules 2020, 25, 3949. [Google Scholar] [CrossRef] [PubMed]
- Raingeval, C.; Cala, O.; Brion, B.; Le Borgne, M.; Hubbard, R.E.; Krimm, I. 1D NMR WaterLOGSY as an efficient method for fragment-based lead discovery. J. Enzym. Inhib. Med. Chem. 2019, 34, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walpole, S.; Monaco, S.; Nepravishta, R.; Angulo, J. STD NMR as a Technique for Ligand Screening and Structural Studies. Methods Enzymol. 2019, 615, 423–451. [Google Scholar] [PubMed]
- Calabrese, D.R.; Connelly, C.M.; Schneekloth, J.S., Jr. Ligand-observed NMR techniques to probe RNA-small molecule interactions. Methods Enzymol. 2019, 623, 131–149. [Google Scholar]
- Hoffmann, M.M.; Sobstyl, H.S.; Badali, V.A. T2 relaxation measurement with solvent suppression and implications to solvent suppression in general. Magn. Reson. Chem. 2009, 47, 593–600. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Olejniczak, E.T.; Fesik, S.W. One-dimensional relaxation- and diffusion-edited NMR methods for screening compounds that bind to macromolecules. J. Am. Chem. Soc. 1997, 119, 12257–12261. [Google Scholar] [CrossRef]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, C.; Zhu, H.; Liu, Z.; Su, H.; Zhang, X.; Chen, T.; Zhong, Y.; Hu, H.; Xiong, M.; et al. Allosteric Regulation of Hsp90alpha’s Activity by Small Molecules Targeting the Middle Domain of the Chaperone. iScience 2020, 23, 100857. [Google Scholar] [CrossRef] [Green Version]
- Cala, O.; Krimm, I. Ligand-Orientation Based Fragment Selection in STD NMR Screening. J. Med. Chem. 2015, 58, 8739–8742. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, J.L.; Taylor, S.L.; Howard, M.J. Recent developments and applications of saturation transfer difference nuclear magnetic resonance (STD NMR) spectroscopy. Mol. Biosyst. 2013, 9, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fogliatto, G.; Stewart, A.; Veronesi, M.; Stockman, B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. Nmr 2001, 21, 349–359. [Google Scholar] [CrossRef]
- Huang, R.; Leung, I.K.H. Protein-Small Molecule Interactions by WaterLOGSY. Methods Enzymol. 2019, 615, 477–500. [Google Scholar] [PubMed]
- Wang, Y.S.; Liu, D.; Wyss, D.F. Competition STD NMR for the detection of high-affinity ligands and NMR-based screening. Magn. Reson. Chem. 2004, 42, 485–489. [Google Scholar] [CrossRef]
- Dalvit, C. Theoretical analysis of the competition ligand-based NMR experiments and selected applications to fragment screening and binding constant measurements. Concepts Magn. Reson. Part A 2008, 32a, 341–372. [Google Scholar] [CrossRef]
- Dalvit, C.; Fagerness, P.E.; Hadden, D.T.; Sarver, R.W.; Stockman, B.J. Fluorine-NMR experiments for high-throughput screening: Theoretical aspects, practical considerations, and range of applicability. J. Am. Chem. Soc. 2003, 125, 7696–7703. [Google Scholar] [CrossRef]
- Vulpetti, A.; Hommel, U.; Landrum, G.; Lewis, R.; Dalvit, C. Design and NMR-Based Screening of LEF, a Library of Chemical Fragments with Different Local Environment of Fluorine. J. Am. Chem. Soc. 2009, 131, 12949–12959. [Google Scholar] [CrossRef]
- Vulpetti, A.; Dalvit, C. Fluorine local environment: From screening to drug design. Drug Discov. Today 2012, 17, 890–897. [Google Scholar] [CrossRef]
- Casale, E.; Amboldi, N.; Brasca, M.G.; Caronni, D.; Colombo, N.; Dalvit, C.; Felder, E.R.; Fogliatto, G.; Galvani, A.; Isacchi, A.; et al. Fragment-based hit discovery and structure-based optimization of aminotriazoloquinazolines as novel Hsp90 inhibitors. Bioorganic Med. Chem. 2014, 22, 4135–4150. [Google Scholar] [CrossRef]
- Dalvit, C.; Ardini, E.; Flocco, M.; Fogliatto, G.P.; Mongelli, N.; Veronesi, M. A general NMR method for rapid, efficient, and reliable biochemical screening. J. Am. Chem. Soc. 2003, 125, 14620–14625. [Google Scholar] [CrossRef] [PubMed]
- Lambruschini, C.; Veronesi, M.; Romeo, E.; Garau, G.; Bandiera, T.; Piomelli, D.; Scarpelli, R.; Dalvit, C. Development of fragment-based n-FABS NMR screening applied to the membrane enzyme FAAH. ChemBioChem 2013, 14, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kang, C. Solution NMR Spectroscopy in Target-Based Drug Discovery. Molecules 2017, 22, 1399. [Google Scholar]
- Fernandez, C.; Jahnke, W. New approaches for NMR screening in drug discovery. Drug Discov. Today: Technol. 2004, 1, 277–283. [Google Scholar] [CrossRef]
- Petros, A.M.; Huth, J.R.; Oost, T.; Park, C.M.; Ding, H.; Wang, X.L.; Zhang, H.C.; Nimmer, P.; Mendoza, R.; Sun, C.H.; et al. Discovery of a potent and selective Bcl-2 inhibitor using SAR by NMR. Bioorg. Med. Chem. Lett. 2010, 20, 6587–6591. [Google Scholar] [CrossRef]
- Chacon Simon, S.; Wang, F.; Thomas, L.R.; Phan, J.; Zhao, B.; Olejniczak, E.T.; Macdonald, J.D.; Shaw, J.G.; Schlund, C.; Payne, W.; et al. Discovery of WD Repeat-Containing Protein 5 (WDR5)-MYC Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2020, 63, 4315–4333. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Li, P.F.; Merz, K.M. Using Ligand-Induced Protein Chemical Shift Perturbations To Determine Protein Ligand Structures. Biochemistry 2017, 56, 2349–2362. [Google Scholar] [CrossRef]
- Francois-Moutal, L.; Scott, D.D.; Perez-Miller, S.; Gokhale, V.; Khanna, M.; Khanna, R. Chemical shift perturbation mapping of the Ubc9-CRMP2 interface identifies a pocket in CRMP2 amenable for allosteric modulation of Nav1.7 channels. Channels 2018, 12, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Fielding, L. NMR methods for the determination of protein-ligand dissociation constants. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 219–242. [Google Scholar] [CrossRef]
- Stadmiller, S.S.; Aguilar, J.S.; Waudby, C.A.; Pielak, G.J. Rapid Quantification of Protein-Ligand Binding via F-19 NMR Lineshape Analysis. Biophys. J. 2020, 118, 2537–2548. [Google Scholar]
- Hajduk, P.J.; Augeri, D.J.; Mack, J.; Mendoza, R.; Yang, J.G.; Betz, S.F.; Fesik, S.W. NMR-based screening of proteins containing C-13-labeled methyl groups. J. Am. Chem. Soc. 2000, 122, 7898–7904. [Google Scholar]
- Gee, C.T.; Koleski, E.J.; Pomerantz, W.C.K. Fragment Screening and Druggability Assessment for the CBP/p300 KIX Domain through Protein-Observed F-19 NMR Spectroscopy. Angew. Chem. Int. Ed. 2015, 54, 3735–3739. [Google Scholar]
- Arntson, K.E.; Pomerantz, W.C. Protein-Observed Fluorine NMR: A Bioorthogonal Approach for Small Molecule Discovery. J. Med. Chem. 2016, 59, 5158–5171. [Google Scholar] [PubMed]
- Mishra, N.K.; Urick, A.K.; Ember, S.W.; Schonbrunn, E.; Pomerantz, W.C. Fluorinated aromatic amino acids are sensitive 19F NMR probes for bromodomain-ligand interactions. ACS Chem. Biol. 2014, 9, 2755–2760. [Google Scholar]
- Orita, M.; Ohno, K.; Warizaya, M.; Amano, Y.; Niimi, T. Lead generation and examples opinion regarding how to follow up hits. Methods Enzymol. 2011, 493, 383–419. [Google Scholar]
- Hopkins, A.L.; Keseru, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar]
- Murray, C.W.; Rees, D.C. Opportunity Knocks: Organic Chemistry for Fragment-Based Drug Discovery (FBDD). Angew. Chem. Int. Ed. Engl. 2016, 55, 488–492. [Google Scholar]
- Mondal, M.; Radeva, N.; Fanlo-Virgos, H.; Otto, S.; Klebe, G.; Hirsch, A.K.H. Fragment Linking and Optimization of Inhibitors of the Aspartic Protease Endothiapepsin: Fragment-Based Drug Design Facilitated by Dynamic Combinatorial Chemistry. Angew. Chem. Int. Ed. 2016, 55, 9422–9426. [Google Scholar]
- Takeuchi, K.; Arthanari, H.; Shimada, I.; Wagner, G. Nitrogen detected TROSY at high field yields high resolution and sensitivity for protein NMR. J. Biomol. Nmr 2015, 63, 323–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, S.; Tailford, L.E.; Juge, N.; Angulo, J. Differential Epitope Mapping by STD NMR Spectroscopy To Reveal the Nature of Protein-Ligand Contacts. Angew. Chem. Int. Ed. Engl. 2017, 56, 15289–15293. [Google Scholar] [CrossRef] [PubMed]
- Nepravishta, R.; Walpole, S.; Tailford, L.; Juge, N.; Angulo, J. Deriving Ligand Orientation in Weak Protein-Ligand Complexes by DEEP-STD NMR Spectroscopy in the Absence of Protein Chemical-Shift Assignment. ChemBioChem 2019, 20, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Friberg, A.; Vigil, D.; Zhao, B.; Daniels, R.N.; Burke, J.P.; Garcia-Barrantes, P.M.; Camper, D.; Chauder, B.A.; Lee, T.; Olejniczak, E.T.; et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013, 56, 15–30. [Google Scholar] [CrossRef]
- Frank, A.O.; Feldkamp, M.D.; Kennedy, J.P.; Waterson, A.G.; Pelz, N.F.; Patrone, J.D.; Vangamudi, B.; Camper, D.V.; Rossanese, O.W.; Chazin, W.J.; et al. Discovery of a potent inhibitor of replication protein a protein-protein interactions using a fragment-linking approach. J. Med. Chem. 2013, 56, 9242–9250. [Google Scholar] [CrossRef] [Green Version]
- Henen, M.A.; Coudevylle, N.; Geist, L.; Konrat, R. Toward rational fragment-based lead design without 3D structures. J. Med. Chem. 2012, 55, 7909–7919. [Google Scholar] [CrossRef]
- Perez, C.; Soler, D.; Soliva, R.; Guallar, V. FragPELE: Dynamic Ligand Growing within a Binding Site. A Novel Tool for Hit-To-Lead Drug Design. J. Chem. Inf. Modeling 2020, 60, 1728–1736. [Google Scholar] [CrossRef]
- Yamaotsu, N.; Hirono, S. In silico fragment-mapping method: A new tool for fragment-based/structure-based drug discovery. J. Comput. Aided Mol. Des. 2018, 32, 1229–1245. [Google Scholar] [CrossRef]
- Sanchez-Pedregal, V.M.; Reese, M.; Meiler, J.; Blommers, M.J.; Griesinger, C.; Carlomagno, T. The INPHARMA method: Protein-mediated interligand NOEs for pharmacophore mapping. Angew. Chem. Int. Ed. Engl. 2005, 44, 4172–4175. [Google Scholar] [CrossRef]
- Seetaha, S.; Yagi-Utsumi, M.; Yamaguchi, T.; Ishii, K.; Hannongbua, S.; Choowongkomon, K.; Kato, K. Application of Site-Specific Spin Labeling for NMR Detecting Inhibitor-Induced Conformational Change of HIV-1 Reverse Transcriptase. ChemMedChem 2016, 11, 363–366. [Google Scholar] [CrossRef]
- Tolman, J.R.; Al-Hashimi, H.M.; Kay, L.E.; Prestegard, J.H. Structural and dynamic analysis of residual dipolar coupling data for proteins. J. Am. Chem. Soc. 2001, 123, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Eletsky, A.; Pulavarti, S.V.S.R.K.; Beaumont, V.; Gollnick, P.; Szyperski, T. Solution NMR Experiment for Measurement of N-15-H-1 Residual Dipolar Couplings in Large Proteins and Supramolecular Complexes. J. Am. Chem. Soc. 2015, 137, 11242–11245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Tjandra, N. The use of residual dipolar coupling in studying proteins by NMR. Top. Curr. Chem. 2012, 326, 47–67. [Google Scholar] [PubMed] [Green Version]
- Pell, A.J.; Pintacuda, G.; Grey, C.P. Paramagnetic NMR in solution and the solid state. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 111, 1–271. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, X.Y.; Lv, G.H.; Razavi, A.M.; Huysmans, G.H.M.; Weinstein, H.; Bracken, C.; Eliezer, D.; Boudker, O. Use of paramagnetic F-19 NMR to monitor domain movement in a glutamate transporter homolog. Nat. Chem. Biol. 2020, 16, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Columbus, L.; Kroncke, B. Solution NMR Structure Determination of Polytopic alpha-Helical Membrane Proteins: A Guide to Spin Label Paramagnetic Relaxation Enhancement Restraints. Membr. Proteins Eng. Purif. Cryst. 2015, 557, 329–348. [Google Scholar]
- Su, X.C.; Otting, G. Paramagnetic labelling of proteins and oligonucleotides for NMR. J. Biomol. NMR 2010, 46, 101–112. [Google Scholar] [CrossRef]
- Wirmer-Bartoschek, J.; Bendel, L.E.; Jonker, H.R.A.; Grun, J.T.; Papi, F.; Bazzicalupi, C.; Messori, L.; Gratteri, P.; Schwalbe, H. Solution NMR Structure of a Ligand/Hybrid-2-G-Quadruplex Complex Reveals Rearrangements that Affect Ligand Binding. Angew. Chem. Int. Ed. Engl. 2017, 56, 7102–7106. [Google Scholar] [CrossRef]
- Tripsianes, K.; Schutz, U.; Emmanouilidis, L.; Gemmecker, G.; Sattler, M. Selective isotope labeling for NMR structure determination of proteins in complex with unlabeled ligands. J. Biomol. Nmr 2019, 73, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Fruth, M.; Plaza, A.; Hinsberger, S.; Sahner, J.H.; Haupenthal, J.; Bischoff, M.; Jansen, R.; Muller, R.; Hartmann, R.W. Binding mode characterization of novel RNA polymerase inhibitors using a combined biochemical and NMR approach. ACS Chem. Biol. 2014, 9, 2656–2663. [Google Scholar] [CrossRef]
- Saio, T.; Ogura, K.; Kumeta, H.; Kobashigawa, Y.; Shimizu, K.; Yokochi, M.; Kodama, K.; Yamaguchi, H.; Tsujishita, H.; Inagaki, F. Ligand-driven conformational changes of MurD visualized by paramagnetic NMR. Sci. Rep. 2015, 5, 16685. [Google Scholar] [CrossRef] [PubMed]
- Goudreau, N.; Hucke, O.; Faucher, A.M.; Grand-Maitre, C.; Lepage, O.; Bonneau, P.R.; Mason, S.W.; Titolo, S. Discovery and structural characterization of a new inhibitor series of HIV-1 nucleocapsid function: NMR solution structure determination of a ternary complex involving a 2:1 inhibitor/NC stoichiometry. J. Mol. Biol. 2013, 425, 1982–1998. [Google Scholar] [CrossRef] [PubMed]
- Davila-Calderon, J.; Patwardhan, N.N.; Chiu, L.Y.; Sugarman, A.; Cai, Z.; Penutmutchu, S.R.; Li, M.L.; Brewer, G.; Hargrove, A.E.; Tolbert, B.S. IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat. Commun. 2020, 11, 4775. [Google Scholar] [CrossRef] [PubMed]
- Petta, I.; Lievens, S.; Libert, C.; Tavernier, J.; De Bosscher, K. Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol. Ther. 2016, 24, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magee, T.V. Progress in discovery of small-molecule modulators of protein-protein interactions via fragment screening. Bioorganic Med. Chem. Lett. 2015, 25, 2461–2468. [Google Scholar] [CrossRef]
- Solbak, S.M.O.; Zang, J.; Narayanan, D.; Hoj, L.J.; Bucciarelli, S.; Softley, C.; Meier, S.; Langkilde, A.E.; Gotfredsen, C.H.; Sattler, M.; et al. Developing Inhibitors of the p47phox-p22phox Protein-Protein Interaction by Fragment-Based Drug Discovery. J. Med. Chem. 2020, 63, 1156–1177. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- De Vries-van Leeuwen, I.J.; da Costa Pereira, D.; Flach, K.D.; Piersma, S.R.; Haase, C.; Bier, D.; Yalcin, Z.; Michalides, R.; Feenstra, K.A.; Jimenez, C.R.; et al. Interaction of 14-3-3 proteins with the estrogen receptor alpha F domain provides a drug target interface. Proc. Natl. Acad. Sci. USA 2013, 110, 8894–8899. [Google Scholar] [CrossRef] [Green Version]
- Ziarek, J.J.; Liu, Y.; Smith, E.; Zhang, G.; Peterson, F.C.; Chen, J.; Yu, Y.; Chen, Y.; Volkman, B.F.; Li, R. Fragment-based optimization of small molecule CXCL12 inhibitors for antagonizing the CXCL12/CXCR4 interaction. Curr. Top. Med. Chem. 2012, 12, 2727–2740. [Google Scholar] [CrossRef] [Green Version]
- Li, C.G.; Liu, M.L. Protein dynamics in living cells studied by in-cell NMR spectroscopy. FEBS Lett. 2013, 587, 1008–1011. [Google Scholar] [PubMed]
- Suemoto, Y.; Tanaka, T.; Kamoshida, H.; Inoue, J.; Mishima, M.; Guentert, P.; Ikeya, T.; Ito, Y. Protein structure determination in living eukaryotic cells by in-cell NMR spectroscopy. Eur. Biophys. J. Biophys. Lett. 2017, 46, S176. [Google Scholar]
- Dotsch, V. Investigation of Proteins in Living Bacteria with in-Cell NMR Experiments. In Bioactive Conformation II; Springer: Cham, Switzerland, 2008; Volume 273, pp. 203–214. [Google Scholar]
- Tanaka, T.; Ikeya, T.; Kamoshida, H.; Suemoto, Y.; Mishima, M.; Shirakawa, M.; Guntert, P.; Ito, Y. High-Resolution Protein 3D Structure Determination in Living Eukaryotic Cells. Angew. Chem. Int. Ed. 2019, 58, 7284–7288. [Google Scholar]
- Barbieri, L.; Luchinat, E.; Banci, L. Characterization of proteins by in-cell NMR spectroscopy in cultured mammalian cells. Nat. Protoc. 2016, 11, 1101–1111. [Google Scholar] [PubMed] [Green Version]
- Sakai, T.; Tochio, H.; Tenno, T.; Ito, Y.; Kokubo, T.; Hiroaki, H.; Shirakawa, M. In-cell NMR spectroscopy of proteins inside Xenopus laevis oocytes. J. Biomol. NMR 2006, 36, 179–188. [Google Scholar] [PubMed]
- Zuberi, Z.; Eeza, M.N.H.; Matysik, J.; Berry, J.P.; Alia, A. NMR-Based Metabolic Profiles of Intact Zebrafish Embryos Exposed to Aflatoxin B1 Recapitulates Hepatotoxicity and Supports Possible Neurotoxicity. Toxins 2019, 11, 258. [Google Scholar]
- Dawes, M.L.; Soeller, C.; Scholpp, S. Studying molecular interactions in the intact organism: Fluorescence correlation spectroscopy in the living zebrafish embryo. Histochem. Cell Biol. 2020, 154, 507–519. [Google Scholar]
- Luchinat, E.; Barbieri, L.; Cremonini, M.; Nocentini, A.; Supuran, C.T.; Banci, L. Drug Screening in Human Cells by NMR Spectroscopy Allows the Early Assessment of Drug Potency. Angew. Chem. Int. Ed. Engl. 2020, 59, 6535–6539. [Google Scholar]
- Burz, D.S.; Shekhtman, A. The STINT-NMR method for studying in-cell protein-protein interactions. Curr. Protoc. Protein Sci. 2010, 61, 17.11.1–17.11.15. [Google Scholar]
- Burz, D.S.; Dutta, K.; Cowburn, D.; Shekhtman, A. Mapping structural interactions using in-cell NMR spectroscopy (STINT-NMR). Nat. Methods 2006, 3, 91–93. [Google Scholar]
- DeMott, C.M.; Girardin, R.; Cobbert, J.; Reverdatto, S.; Burz, D.S.; McDonough, K.; Shekhtman, A. Potent Inhibitors of Mycobacterium tuberculosis Growth Identified by Using in-Cell NMR-based Screening. ACS Chem. Biol. 2018, 13, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Barbieri, L.; Cremonini, M.; Nocentini, A.; Supuran, C.T.; Banci, L. Intracellular Binding/Unbinding Kinetics of Approved Drugs to Carbonic Anhydrase II Observed by in-Cell NMR. ACS Chem. Biol. 2020, 15, 2792–2800. [Google Scholar] [CrossRef] [PubMed]
- Primikyri, A.; Sayyad, N.; Quilici, G.; Vrettos, E.I.; Lim, K.; Chi, S.W.; Musco, G.; Gerothanassis, I.P.; Tzakos, A.G. Probing the interaction of a quercetin bioconjugate with Bcl-2 in living human cancer cells with in-cell NMR spectroscopy. FEBS Lett. 2018, 592, 3367–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potenza, D.; Vasile, F.; Belvisi, L.; Civera, M.; Araldi, E.M. STD and trNOESY NMR study of receptor-ligand interactions in living cancer cells. ChemBioChem 2011, 12, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Orts, J.; Riek, R. Protein-ligand structure determination with the NMR molecular replacement tool, NMR(2). J. Biomol. NMR 2020, 74, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Fino, R.; Byrne, R.; Softley, C.A.; Sattler, M.; Schneider, G.; Popowicz, G.M. Introducing the CSP Analyzer: A novel Machine Learning-based application for automated analysis of two-dimensional NMR spectra in NMR fragment-based screening. Comput. Struct. Biotechnol. J. 2020, 18, 603–611. [Google Scholar] [CrossRef]
- Strotz, D.; Orts, J.; Chi, C.N.; Riek, R.; Vogeli, B. eNORA2 Exact NOE Analysis Program. J. Chem. Theory Comput. 2017, 13, 4336–4346. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Breton, R.C.; Burns, D.C. Evaluating ASAP-HMQC and PS-HSQC NMR pulse sequences with non-uniform sampling for rapid screening of natural products. Planta Med. 2014, 80, IL26. [Google Scholar] [CrossRef]
- Codutti, L.; Grimaldi, M.; Carlomagno, T. Structure-Based Design of Scaffolds Targeting PDE10A by INPHARMA-NMR. J. Chem. Inf. Modeling 2017, 57, 1488–1498. [Google Scholar] [CrossRef]
- Sikorska, J.; Codutti, L.; Ameneiro, R.S.; Skjaerven, L.; Angelini, A.; Carlomagno, T. Understanding of Protein-Ligand Interactions with INPHARMA. Planta Med. 2013, 79, PD2. [Google Scholar] [CrossRef]
- Mateos, B.; Konrat, R.; Pierattelli, R.; Felli, I.C. NMR Characterization of Long-Range Contacts in Intrinsically Disordered Proteins from Paramagnetic Relaxation Enhancement in C-13 Direct-Detection Experiments. ChemBioChem 2019, 20, 335–339. [Google Scholar] [CrossRef]
- Nishida, N.; Ito, Y.; Shimada, I. In situ structural biology using in-cell NMR. Biochim. Biophysca Acta Gen. Subj. 2020, 1864, 129364. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Banci, L. In-cell NMR: A topical review. IUCrJ 2017, 4, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airoldi, C.; Giovannardi, S.; La Ferla, B.; Jimenez-Barbero, J.; Nicotra, F. Saturation transfer difference NMR experiments of membrane proteins in living cells under HR-MAS conditions: The interaction of the SGLT1 co-transporter with its ligands. Chemistry 2011, 17, 13395–13399. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.S.; Liu, X.L.; Zhang, Z.T.; Wu, Q.; Jiang, B.; Jiang, L.; Zhang, X.; Liu, M.L.; Pielak, G.J.; Li, C.G. F-19 NMR Spectroscopy as a Probe of Cytoplasmic Viscosity and Weak Protein Interactions in Living Cells. Chem. Eur. J. 2013, 19, 12705–12710. [Google Scholar] [CrossRef]
- Li, C.G.; Wang, G.F.; Wang, Y.Q.; Creager-Allen, R.; Lutz, E.A.; Scronce, H.; Slade, K.M.; Ruf, R.A.S.; Mehl, R.A.; Pielak, G.J. Protein F-19 NMR in Escherichia coli. J. Am. Chem. Soc. 2010, 132, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Kubo, S.; Nishida, N.; Udagawa, Y.; Takarada, O.; Ogino, S.; Shimada, I. A Gel-Encapsulated Bioreactor System for NMR Studies of Protein-Protein Interactions in Living Mammalian Cells. Angew. Chem. Int. Ed. 2013, 52, 1208–1211. [Google Scholar] [CrossRef]
- Sharaf, N.G.; Barnes, C.O.; Charlton, L.M.; Young, G.B.; Pielak, G.J. A bioreactor for in-cell protein NMR. J. Magn. Reson. 2010, 202, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Motta, A.; Paris, D.; Melck, D. Monitoring Real-Time Metabolism of Living Cells by Fast Two-Dimensional NMR Spectroscopy. Anal. Chem. 2010, 82, 2405–2411. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Techniques | Advantages | Disadvantages |
|---|---|---|
| Thermal Shift Assay (TSA) | Low cost and high throughput; Identification of hit compounds which modify the target’s thermal stability [68]. | False positive results are possible. |
| Surface Plasmon Resonance (SPR) | High throughput; Detection of binding interactions in real time and in a label-free manner [79]. | Targets need to be immobilized; False positive results are possible. |
| Weak Affinity Chromatography-Mass Spectroscopy (WAC-MS) | High sensitivity; Label free; High efficiency [80]. | False positive results are possible [81,82]. |
| X-ray Crystallography | Capable of providing high-resolution and detailed structural information for binding interactions | High quality target crystals are needed. |
| MicroScale Thermophoresis (MST) | Low cost; high throughput and high efficiency [69]. | Fluorescent labeling is needed; False positive results are possible [69]. |
| Ligand-observed NMR | No molecular weight limitation for targets; Capable of detecting weak binders. | False positive results are possible due to compound aggregation. |
| Target-observed NMR | Capable of providing binding site information; Capable of detecting weak binders. | Isotope labeling is needed; Low throughput; Molecular weight limitation for targets. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Zhang, N. Applications of Solution NMR in Drug Discovery. Molecules 2021, 26, 576. https://doi.org/10.3390/molecules26030576
Shi L, Zhang N. Applications of Solution NMR in Drug Discovery. Molecules. 2021; 26(3):576. https://doi.org/10.3390/molecules26030576
Chicago/Turabian StyleShi, Li, and Naixia Zhang. 2021. "Applications of Solution NMR in Drug Discovery" Molecules 26, no. 3: 576. https://doi.org/10.3390/molecules26030576
APA StyleShi, L., & Zhang, N. (2021). Applications of Solution NMR in Drug Discovery. Molecules, 26(3), 576. https://doi.org/10.3390/molecules26030576