
New 1,2,3-Triazole-Containing Hybrids as Antitumor Candidates: Design, Click Reaction Synthesis, DFT Calculations, and Molecular Docking Study



Abstract
:
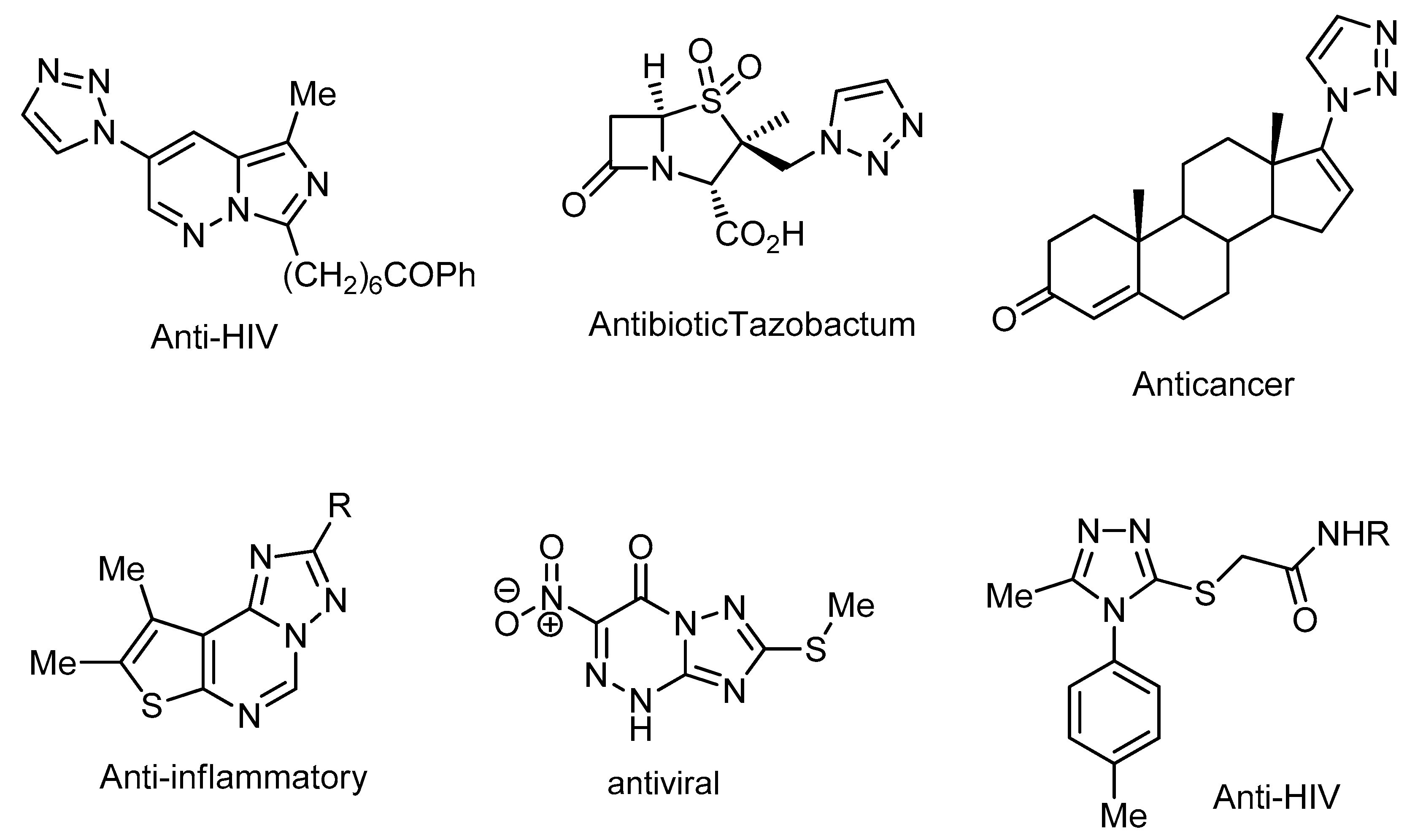
1. Introduction
2. Results and Discussion
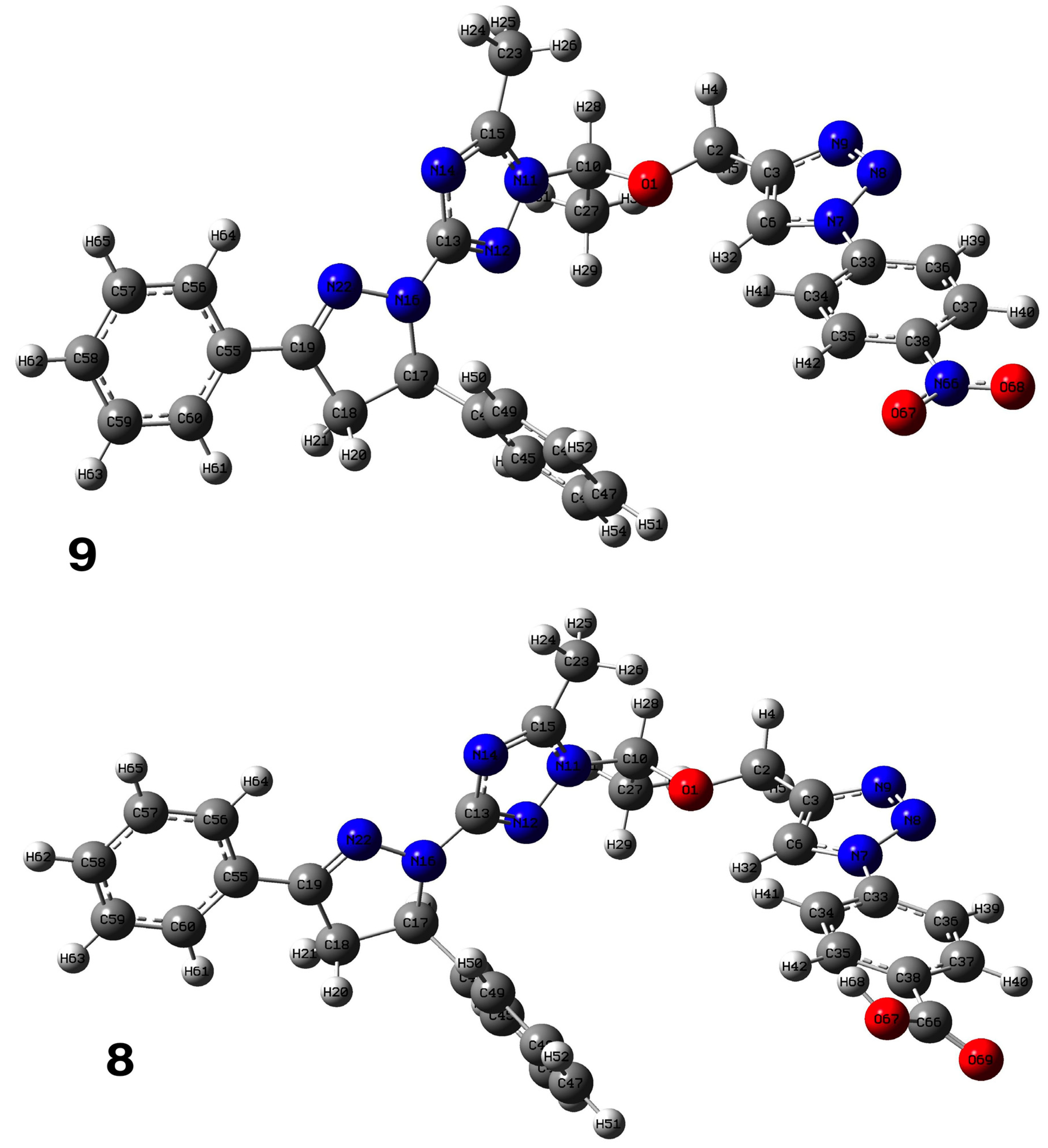
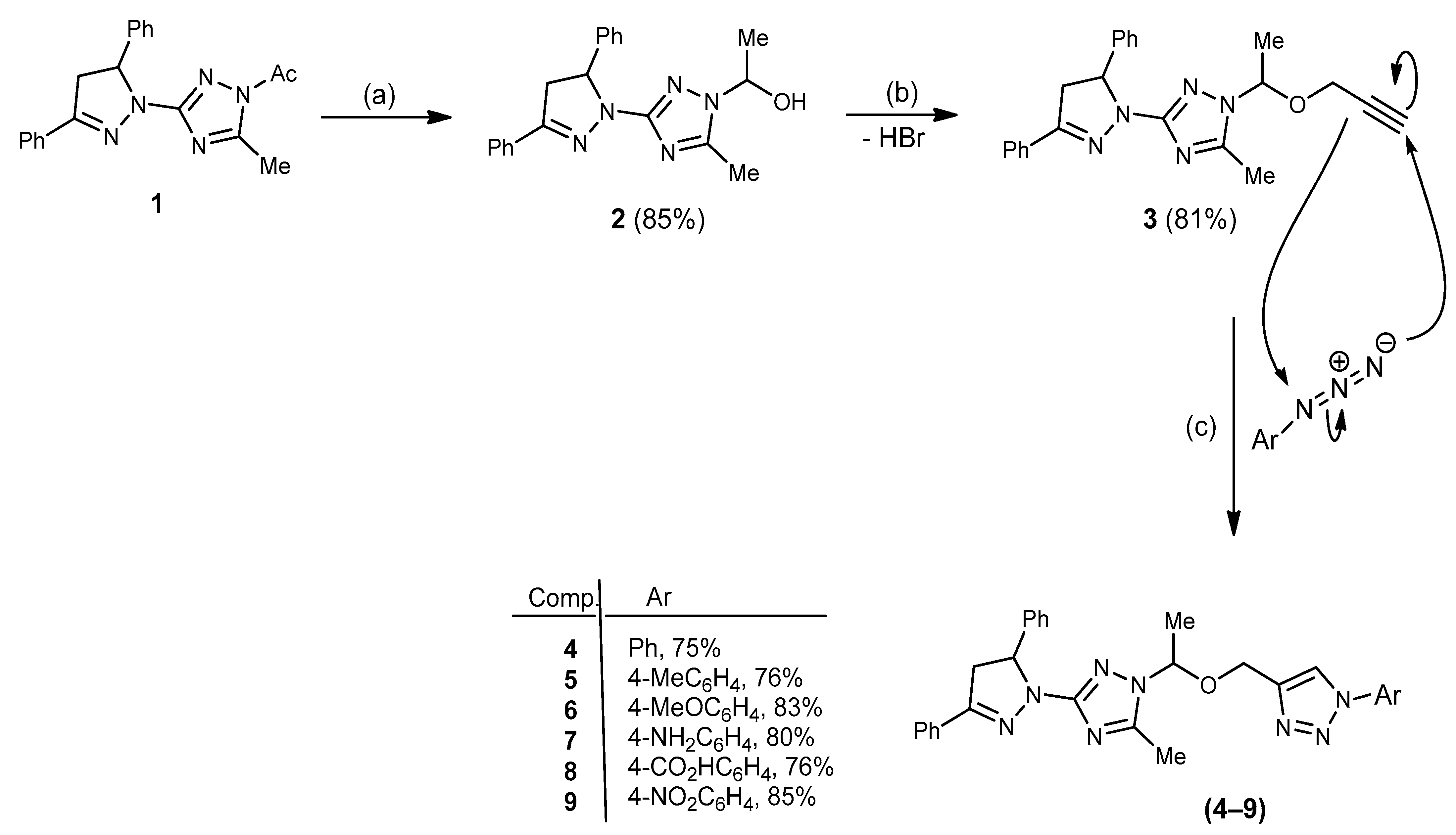
2.1. Chemistry
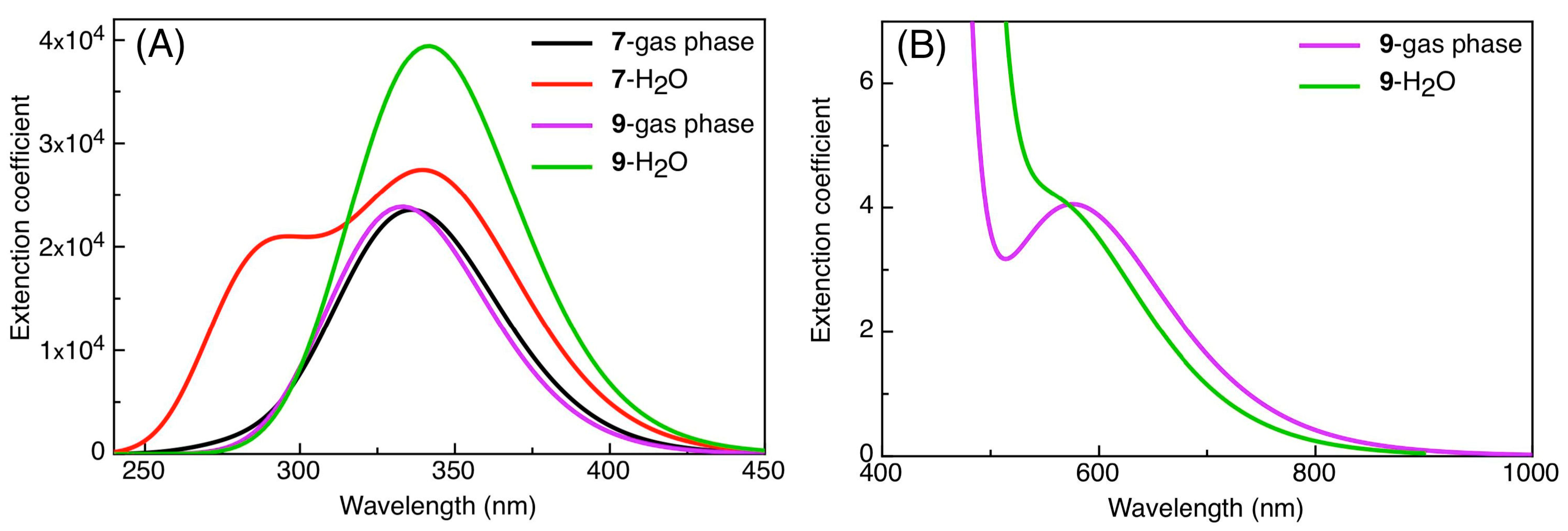
2.2. DFT Calculations
2.3. Pharmacological Evaluation
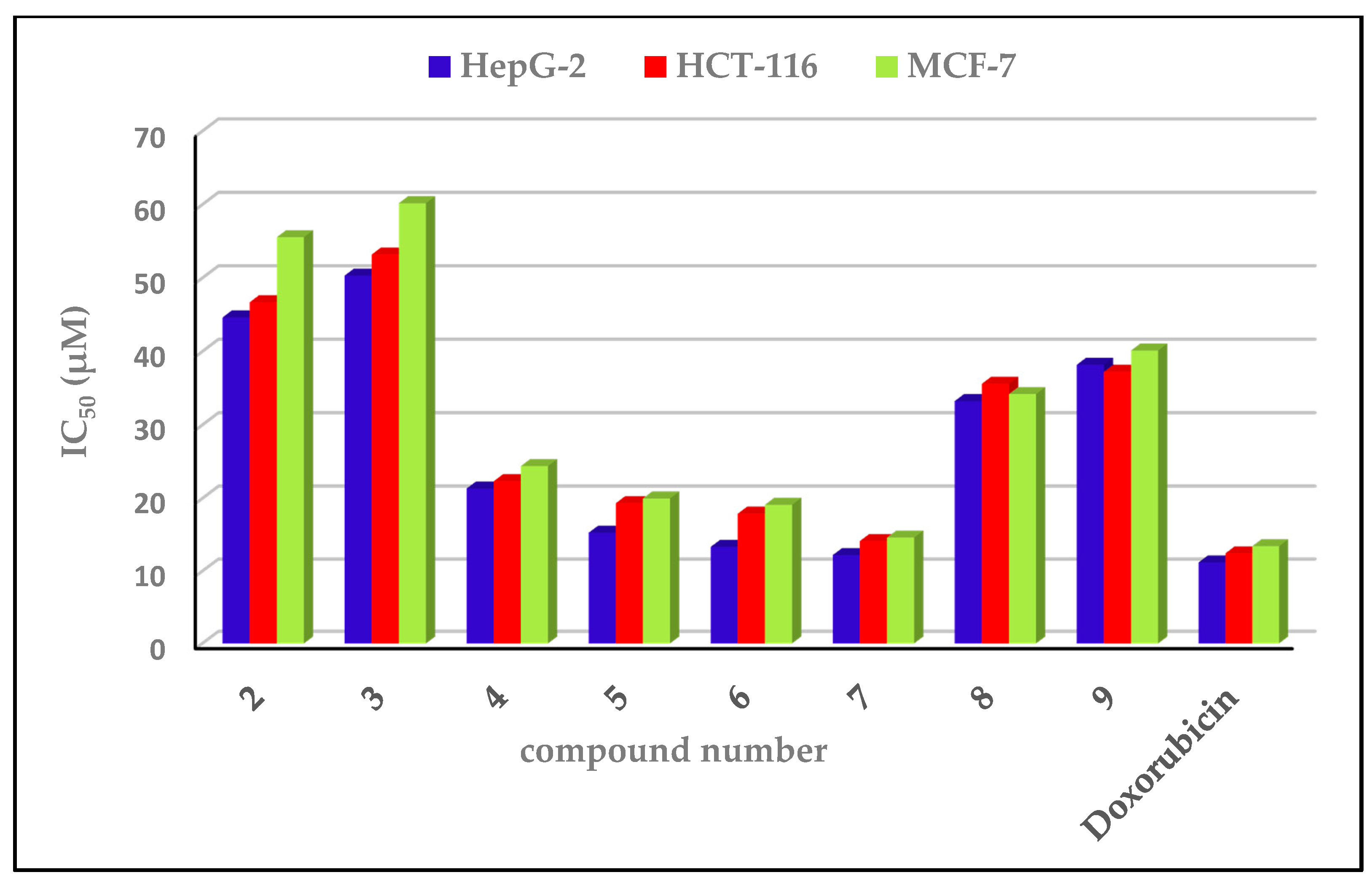
2.3.1. Cytotoxic Impacts
2.3.2. Structure–Activity Relationship
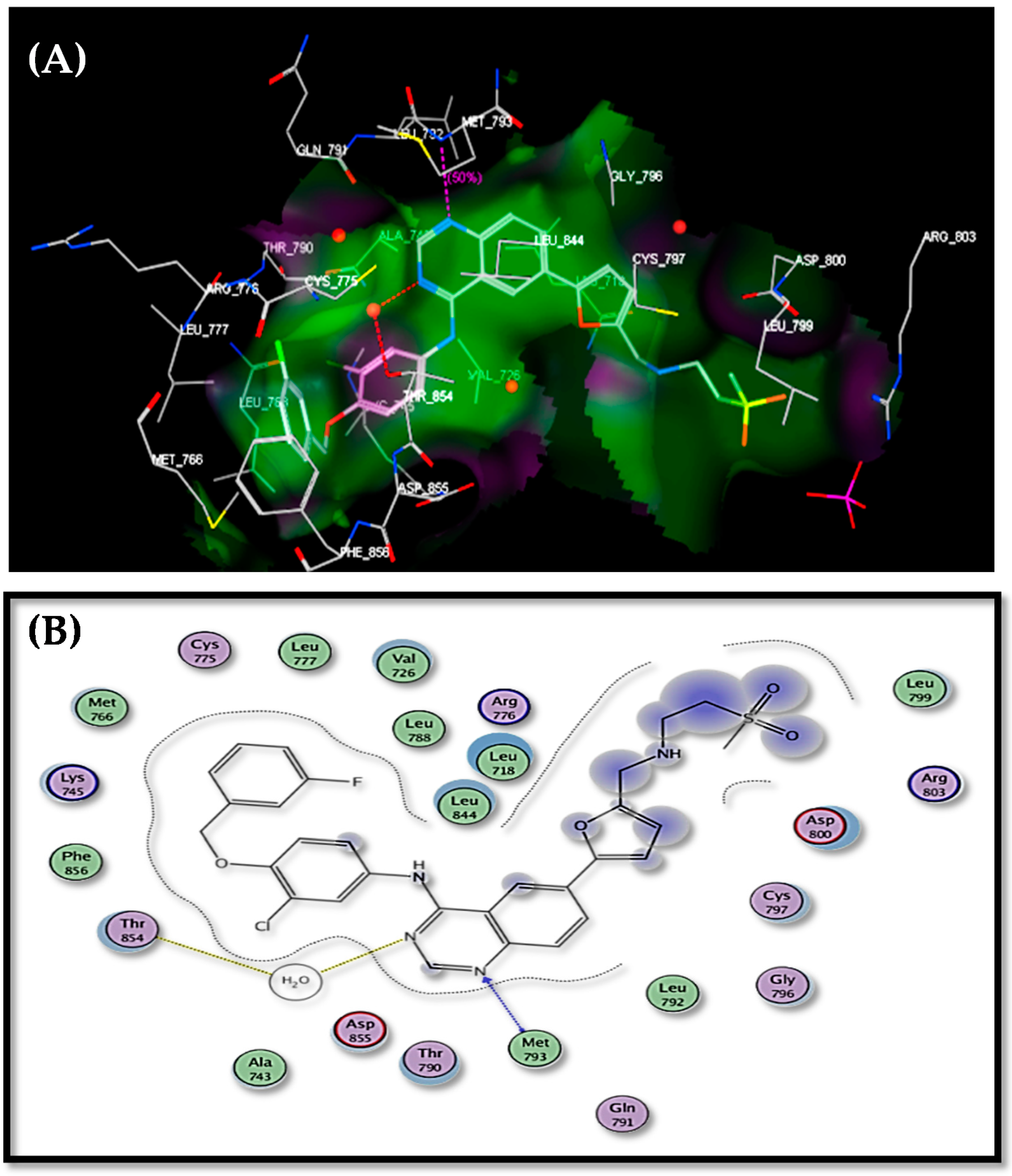
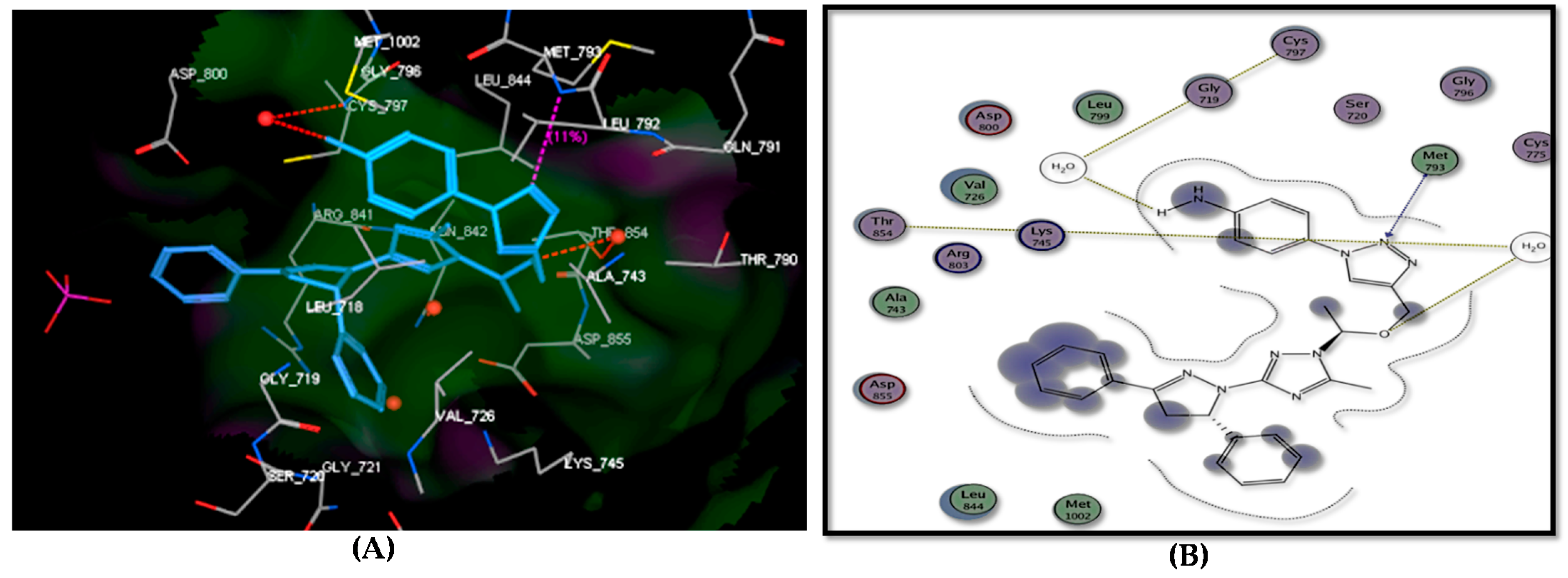
2.4. Docking Study
3. Materials and Methods
3.1. General Description of Materials and Methods
3.2. Instrumentation
3.3. Synthetic Procedures and Analytic Data of Compounds
3.4. Cytotoxic Assessment
Methodology
3.5. Molecular Docking Study
3.6. DFT Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sangthong, S.; Krusong, K.; Ngamrojanavanich, N.; Vilaivan, T.; Puthong, S.; Chandchawan, S.; Muangsin, N. Synthesis of rotenoid derivatives with cytotoxic and topoisomerase II inhibitory activities. Bioorg. Med. Chem. Lett. 2011, 21, 4813–4818. [Google Scholar] [CrossRef]
- Popiołek, Ł.; Biernasiuk, A.; Paruch, K.; Malm, A.; Wujec, M. Synthesis and in vitro antimicrobial activity screening of new pipemidic acid derivatives. Arch. Pharm. Res. 2018, 41, 633–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Zhao, S.-J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700–111713. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.; Gomaa, M.S.; El Sayed, H.; Duerkop, A. Design, selective alkylation and X-ray crystal structure determination of dihydro-indolyl-1,2,4-triazole-3-thione and its 3-benzylsulfanyl analogue as potent anticancer agents. Eur. J. Med. Chem. 2017, 125, 360–371. [Google Scholar] [CrossRef]
- Aouad, M.R.; Mayaba, M.M.; Naqvi, A.; Bardaweel, S.K.; Al-blewi, F.F.; Messali, M.; Rezki, N. Design, synthesis, in silico and in vitro antimicrobial screenings of novel 1,2,4-triazoles carrying 1,2,3-triazole scaffold with lipophilic side chain tether. Chem. Cent. J. 2017, 11, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timur, I.; Kocyigit, Ü.M.; Dastan, T.; Sandal, S.; Ceribası, A.O.; Taslimi, P.; Gulcin, ˙I.; Koparir, M.; Karatepe, M.; Çiftçi, M.J. In vitro cytotoxic and in vivo antitumoral activities of some aminomethyl derivatives of 2,4-dihydro-3H-1,2,4-triazole-3-thiones-Evaluation of their acetylcholinesterase and carbonic anhydrase enzymes inhibition profiles. Biochem. Mol. Toxicol. 2019, 33, 22239–22250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapro’n, B.; Łuszczki, J.J.; Płazi´nska, A.; Siwek, A.; Karcz, T.; Grybo´s, A.; Nowak, G.; Makuch-Kocka, A.; Walczak, K.; Langner, E. Development of the 1,2,4-triazole-based anticonvulsant drug candidates acting on the voltage-gated sodium channels. Insights from in-vivo, in-vitro, and in-silico studies. Eur. J. Pharm. Sci. 2019, 129, 42–57. [Google Scholar] [CrossRef]
- Savanur, H.M.; Naik, K.N.; Ganapathi, S.M.; Kim, K.M.; Kalkhambkar, R.G. Click chemistry inspired design, synthesis and molecular docking studies of coumarin, quinolinone linked 1,2,3-triazoles as promising anti-microbial agents. Chem. Select 2018, 3, 5296–5303. [Google Scholar] [CrossRef]
- Rajavelu, K.; Subaraja, M.; Rajakumar, P. Synthesis, optical properties, and antioxidant and anticancer activity of benzoheterazole dendrimers with triazole bridging unit. New J. Chem. 2018, 42, 3282–3292. [Google Scholar] [CrossRef]
- Kumar, K.A.; Kalluraya, B.; Kumar, S.M. Synthesis and in-vitro antioxidant activities of some coumarin derivatives containing 1,2,3-triazole ring. Phosphorus Sulfur Silicon Relat. Elem. 2018, 193, 294–299. [Google Scholar] [CrossRef]
- Santosh, R.; Selvam, M.K.; Kanekar, S.U.; Nagaraja, G.K. Synthesis, characterization, antibacterial and antioxidant studies of some heterocyclic compounds from triazolelinked chalcone derivatives. Chem. Select 2018, 3, 6338–6343. [Google Scholar] [CrossRef]
- Golas, P.L.; Matyjaszewski, K. Click Chemistry and ATRP: A Beneficial Union for the Preparation of Functional Materials. QSAR Comb. Sci. 2007, 26, 1116–1134. [Google Scholar] [CrossRef]
- Nuzzi, A.; Massi, A.; Dondoni, A. Model Studies Toward the Synthesis of Thymidine Oligonucleotides with Triazole Internucleosidic Linkages Via Iterative Cu(I)-Promoted Azide–Alkyne Ligation Chemistry. QSAR Comb. Sci. 2007, 26, 1191–1199. [Google Scholar] [CrossRef]
- Gierlich, J.; Burley, G.A.; Gramlich, P.M.E.; Hammond, D.M.; Carell, T. Click Chemistry as a Reliable Method for the High-Density PostsyntheticFunctionalization of Alkyne-Modified DNA. Org. Lett. 2006, 8, 3639–3642. [Google Scholar] [CrossRef]
- Savaş, B.; Öztürk, T.; Meyvacı, E.; Hazer, B. Synthesis and characterization of comb-type graft copolymers by redox polymerization and “click” chemistry method. SN Appl. Sci. 2020, 2, 18–26. [Google Scholar] [CrossRef] [Green Version]
- El Malah, T.; Nour, H.F.; Satti, A.A.E.; Hemdan, B.A.; El-Sayed, W.A. Design, Synthesis, and Antimicrobial Activities of 1,2,3-Triazole Glycoside Clickamers. Molecules 2020, 25, 790. [Google Scholar] [CrossRef] [Green Version]
- Kollaschinski, M.; Sobotta, J.; Schalk, A.; Frischmuth, T.; Graf, B.; Serdjukow, S. Efficient DNA Click Reaction Replaces Enzymatic Ligation. Bioconj. Chem. 2020, 31, 507–512. [Google Scholar] [CrossRef]
- Samarasimhareddy, M.; Shamir, M.; Shalev, D.E.; Hurevich, M.; Friedler, A.A. Rapid and Efficient Building Block Approach for Click Cyclization of Peptoids. Front. Chem. 2020, 8, 405–420. [Google Scholar] [CrossRef]
- Yucheng, Z.; Chu, C.-W.; Wei, M.; Atsushi, T. Functionalization of Metal Surface via Thiol–Ene Click Chemistry: Synthesis, Adsorption Behavior, and Postfunctionalization of a Catechol- and Allyl-Containing Copolymer. ACS Omega 2020, 5, 7488–7496. [Google Scholar] [CrossRef]
- Stefanetti, G.; Allan, M.; Usera, A.; Micoli, F. Click chemistry compared to thiol chemistry for the synthesis of site-selective glycoconjugate vaccines using CRM197 as carrier protein. Glycoconj. J. 2020, 37, 611–622. [Google Scholar] [CrossRef]
- El Azab, I.H.; Elkanzi, N.A.A. Design, Synthesis, and Antimicrobial Evaluation of New Annelated Pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazines. Molecules 2020, 25, 1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Azab, I.H.; Elkanzi, N.A.A. An Efficient Synthetic Approach Towards Benzo[b]pyrano[2,3-e][1,4]diazepines and Their Cytotoxic Activity. Molecules 2020, 25, 2051. [Google Scholar] [CrossRef] [PubMed]
- El Azab, I.H.; Elkanzi, N.A.A.; Gobouri, A.A. Design and Synthesis of Some New Quinoxaline-Based Heterocycles, J. Heterocycl. Chem. 2018, 55, 65–76. [Google Scholar] [CrossRef]
- El Azab, I.H.; Gobouri, A.A.; Altalhi, T.A. 4-Chlorothiazole-5-carbaldehydes as Potent Precursors for Synthesis of Some New Pendant N-heterocyces Endowed with Anti-Tumor Activity. J. Heterocycl. Chem. 2019, 56, 281–295. [Google Scholar] [CrossRef] [Green Version]
- El Azab, I.H.; Elkanzi, N.A.A.; Gobouri, A.A.; Altalhi, T.A. Convenient Synthesis of Novel Nitrogen Bridgehead Heterocycles Utilizing 3-Mercapto-6H-[1,2,4,5]oxatriazino[3,2-a] isoindol-6-one as a New Synthon. J. Heterocycl. Chem. 2019, 56, 60–72. [Google Scholar] [CrossRef] [Green Version]
- El Azab, I.H.; Abu Ali, O.A.; El-Zahrani, A.H.; Gobouri, A.A.; Altalhi, T.A. Pyrazole-1-carbothioamide as a Potent Precursor for Synthesis of Some New N-heterocycles of Potential Biological Activity. J. Heterocycl. Chem. 2019, 56, 18–31. [Google Scholar] [CrossRef] [Green Version]
- El Azab, I.H.; Khalifa, M.E.; Gobouri, A.A.; Altalhi, T.A. Synthesis, Characterization, and Pharmacological Evaluation of Some New Pteridine-Based Heterocycles as Antimicrobial Agents. J. Heterocycl. Chem. 2019, 56, 1352–1361. [Google Scholar] [CrossRef]
- El-Sheshtawy, H.S.; Abou Baker, A.M. Synthesis, structural, theoretical studies and biological activities of 3-(arylamino)-2-phenyl-1H-inden-1-one derivative. J. Mol. Struct. 2014, 1067, 225–232. [Google Scholar] [CrossRef]
- El-Sheshtawy, H.S.; Abdelmonsef, A.H.; Abboudy, S.M.; Younes, A.M.M.; Taha, M.M.; Hassan, M.A. Synthesis, Structural, and Theoretical Studies of Quinazoline-2,4-dione Derivatives. Polycycl. Aromat. Compd. 2019, 39, 279–286. [Google Scholar] [CrossRef]
- El-Sheshtawy, H.S.; Assran, A.S.; AbouBaker, A.M. Synthesis, Structural Characterization, Spectroscopic Properties, and Theoretical Investigations of Aminoacridine Derivatives. Polycycl. Aromat. Compd. 2019, 39, 1–13. [Google Scholar] [CrossRef]
- Youns, M.; Fu, Y.J.; Zu, Y.G.; Kramer, A.; Konkimalla, V.B.; Radlwimmer, B.; Sultmann, H.; Efferth, T. Sensitivity and resistance towards isoliquiritigenin, doxorubicin and methotrexate in T cell acute lymphoblastic leukaemia cell lines by pharmacogenomics. Arch. Pharmacol. 2010, 382, 221–234. [Google Scholar] [CrossRef]
- Kuete, V.; Krusche, B.; Youns, M.; Voukeng, I.; Fankam, A.G.; Tankeo, S.; Lacmata, S.; Efferth, T. Cytotoxicity of some Cameroonian spices and selected medicinal plant extracts. J. Ethnopharmacol. 2011, 134, 803–812. [Google Scholar] [CrossRef]
- Schlessinger, J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004, 306, 1506–1507. [Google Scholar] [CrossRef]
- Wang, Y.N.; Yamaguchi, H.; Hsu, J.; Hung, M. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010, 29, 3997–4006. [Google Scholar] [CrossRef] [Green Version]
- Gschwind, A.; Zwick, E.; Prenzel, N.; Leserer, M.; Ullrich, A. Cell communication networks: Epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 2001, 20, 1594–1600. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, 3–8. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Alkhoja, O.A.; Mohamed, W.R. Design, synthesis and antitumor activity of novel pyrazolo[3,4-d]pyrimidine derivatives as EGFR-TK inhibitors. Bioorg. Chem. 2016, 66, 88–96. [Google Scholar] [CrossRef]
- Bakr, R.B.; Mehany, A.B.M.; Abdellatif, K.R.A. Synthesis, EGFR Inhibition and Anti-cancer Activity of New 3,6-dimethyl-1-phenyl-4-(substituted-methoxy)pyrazolo[3,4-d]pyrimidine Derivatives. Curr. Med. Chem. Anti Cancer Agents 2017, 17, 1389–1400. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision d1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. No | HOMO | LUMO |
|---|---|---|
| 4 |  |  |
| 5 |  |  |
| 6 |  |  |
| 7 |  |  |
| 8 |  |  |
| 9 |  |  |
| Comp. | EHOMO (eV) | ELUMO (eV) | Eg (eV) | λmax (nm) | f |
|---|---|---|---|---|---|
| 4 | −5.43 (−5.68) a | −1.42 (−1.61) | 4.01 (4.07) | 334 (341) | 0.58 |
| 5 | −5.42 (−5.69) | −1.35 (−1.62) | 4.07 (4.07) | 336 (341) | 0.58 |
| 6 | −5.30 (−5.66) | −1.26 (−1.60) | 4.04 (4.06) | 340 (343) | 0.59 |
| 7 | −5.39 (−5.67) | −1.32 (−1.61) | 4.07 (4.06) | 337 (291, 341) | 0.58 |
| 8 | −5.54 (−5.68) | −2.34 (−2.26) | 3.20 (3.42) | 333 (341) | 0.59 |
| 9 | −5.55 (−5.68) | −3.13 (−3.21) | 2.42 (2.47) | 333, 580 (343, 585) | 0.58 |
| Compound No. | IC50 (µM) ± SD | ||
|---|---|---|---|
| HepG-2 | HCT-116 | MCF-7 | |
| 2 | 44.65 ± 2.03 | 46.71 ± 1.35 | 55.57 ± 1.89 |
| 3 | 50.34 ± 1.18 | 53.21 ± 0.31 | 60.20 ± 0.36 |
| 4 | 21.35 ± 0.67 | 22.35 ± 0.96 | 24.41 ± 0.36 |
| 5 | 15.31 ± 0.65 | 19.35 ± 1.41 | 20.00 ± 0.46 |
| 6 | 13.36 ± 0.24 | 17.93 ± 0.53 | 19.14± 0.87 |
| 7 | 12.22 ± 1.02 | 14.16 ± 0.38 | 14.64 ± 0.64 |
| 8 | 33.26 ± 0.62 | 35.61 ± 1.21 | 34.21 ± 1.01 |
| 9 | 38.20 ± 1.61 | 37.24 ± 0.32 | 40.14 ± 1.05 |
| Doxorubicin | 11.21± 0.14 | 12.46 ± 0.19 | 13.45 ± 0.54 |
| Target No. | E. Score Kcal/mol | hydrogen Bonds Number | Distance (Ao) from Amino Acid | Binded Group | |
|---|---|---|---|---|---|
| 2 | −14.50 | 1 | 3.05 | Met793 | OH |
| 3 | −14.34 | 1 | 2.98 | Cys797 | N-2 of pyrazole |
| 4 | −16.22 | 1 | 2.82 | Thr854 | N-2 of triazole |
| 5 | −15.83 | 2 | 3.54 2.75 | Thr854 (via H2O) Lys745 | N-2 of triazole OCH2 |
| 6 | −16.70 | 2 | 2.83 2.59 | Thr854 (via H2O) Lys745 | OMe N-2 of triazole |
| 7 | −17.01 | 3 | 2.44 2.48 2.97 | Thr854 (via H2O) Met793 Cys797 | OCH2 N-2 of triazole NH2 |
| 8 | −15.02 | 2 | 3.22 2.98 | Cys797 (via H2O) Thr854 (via H2O) | N-3 of triazole CO2H |
| 9 | −14.67 | 1 | 2.79 | Thr854 | N-2 of pyrazole |
| Lapatinib | −17.22 | 2 | 2.30 2.41 | Thr854 (via H2O) Met793 | N-3 of quinazoline N-1 of quinazoline |
Sample Availability: Samples of compounds 2–9 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Azab, I.H.; El-Sheshtawy, H.S.; Bakr, R.B.; Elkanzi, N.A.A. New 1,2,3-Triazole-Containing Hybrids as Antitumor Candidates: Design, Click Reaction Synthesis, DFT Calculations, and Molecular Docking Study. Molecules 2021, 26, 708. https://doi.org/10.3390/molecules26030708
El Azab IH, El-Sheshtawy HS, Bakr RB, Elkanzi NAA. New 1,2,3-Triazole-Containing Hybrids as Antitumor Candidates: Design, Click Reaction Synthesis, DFT Calculations, and Molecular Docking Study. Molecules. 2021; 26(3):708. https://doi.org/10.3390/molecules26030708
Chicago/Turabian StyleEl Azab, Islam H., Hamdy S. El-Sheshtawy, Rania B. Bakr, and Nadia A. A. Elkanzi. 2021. "New 1,2,3-Triazole-Containing Hybrids as Antitumor Candidates: Design, Click Reaction Synthesis, DFT Calculations, and Molecular Docking Study" Molecules 26, no. 3: 708. https://doi.org/10.3390/molecules26030708
APA StyleEl Azab, I. H., El-Sheshtawy, H. S., Bakr, R. B., & Elkanzi, N. A. A. (2021). New 1,2,3-Triazole-Containing Hybrids as Antitumor Candidates: Design, Click Reaction Synthesis, DFT Calculations, and Molecular Docking Study. Molecules, 26(3), 708. https://doi.org/10.3390/molecules26030708