
A Contribution to the Solid State Forms of Bis(demethoxy)curcumin: Co-Crystal Screening and Characterization



Abstract
:1. Introduction
2. Results and Discussion
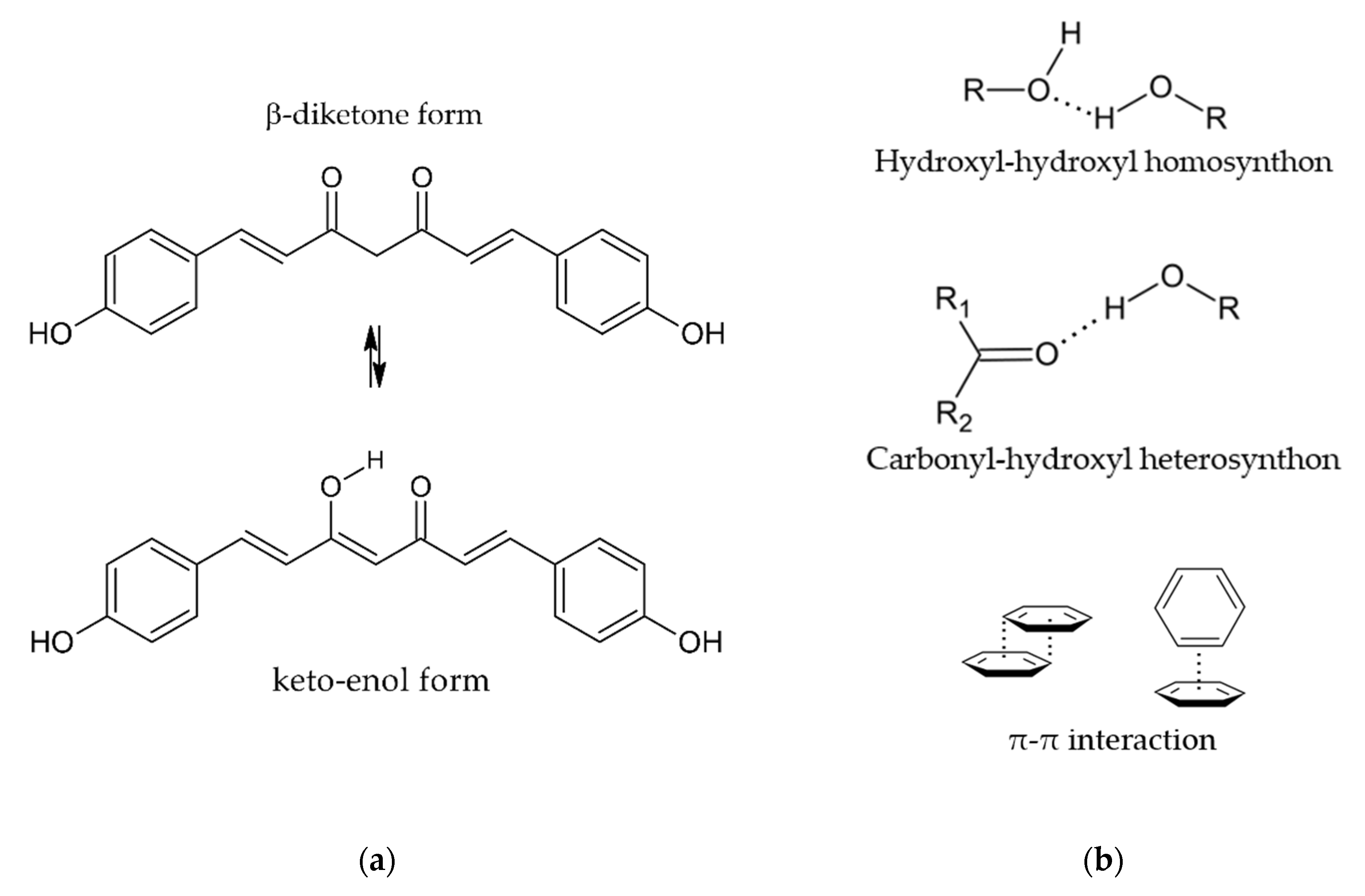
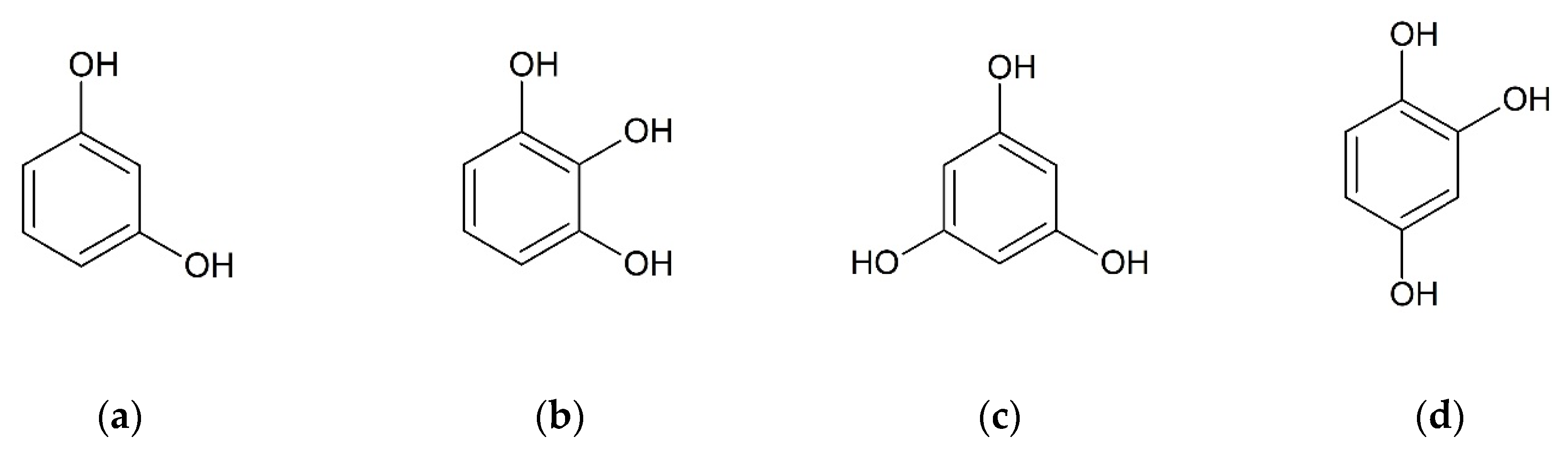
2.1. Selection of Coformers
- hydroxyl-hydroxyl homosynthon
- carbonyl-hydroxyl heterosynthon
- π-π interaction
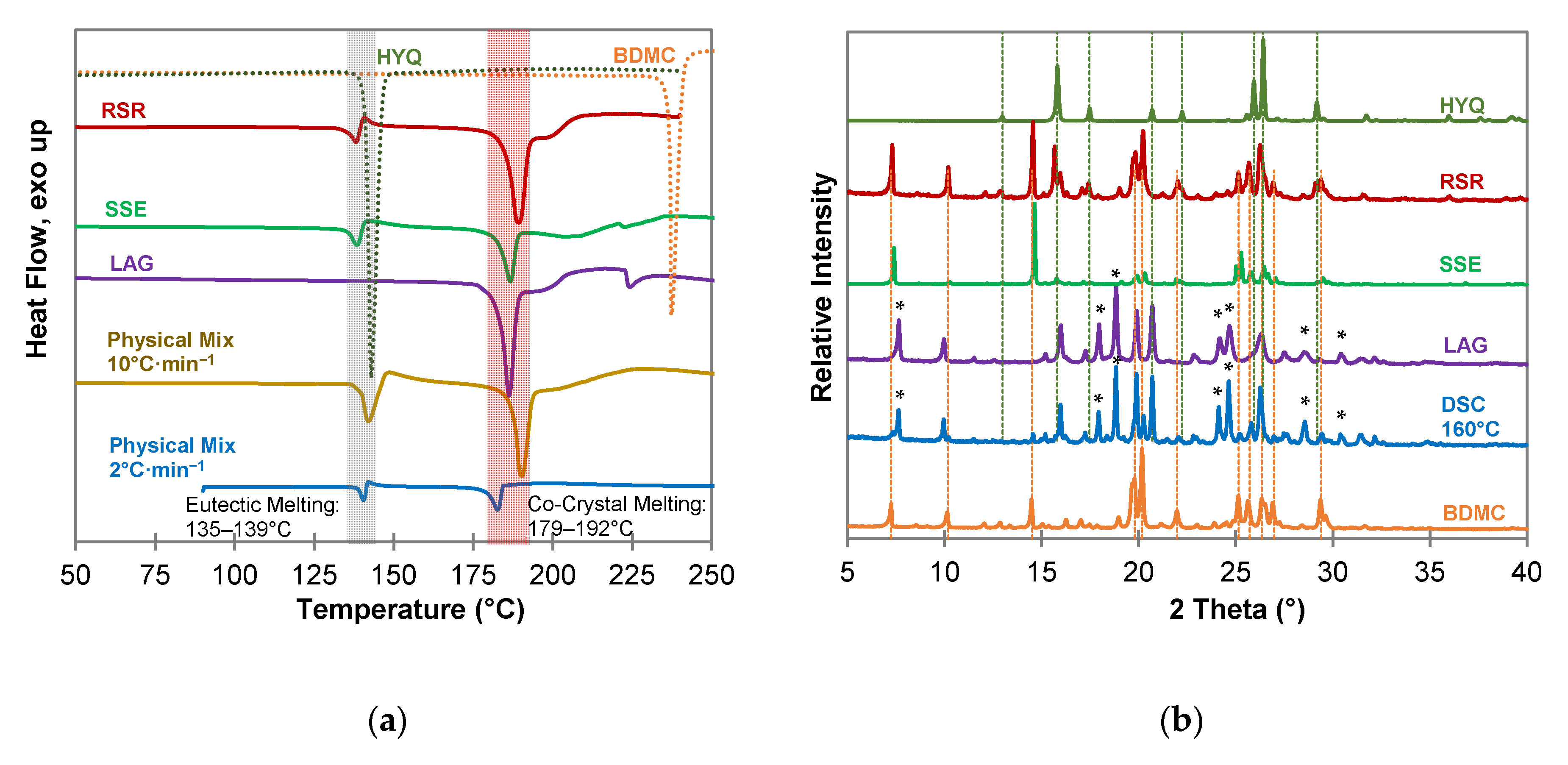
2.2. Co-Crystallization of Bis(demethoxy)curcumin with Hydroxybenzenes
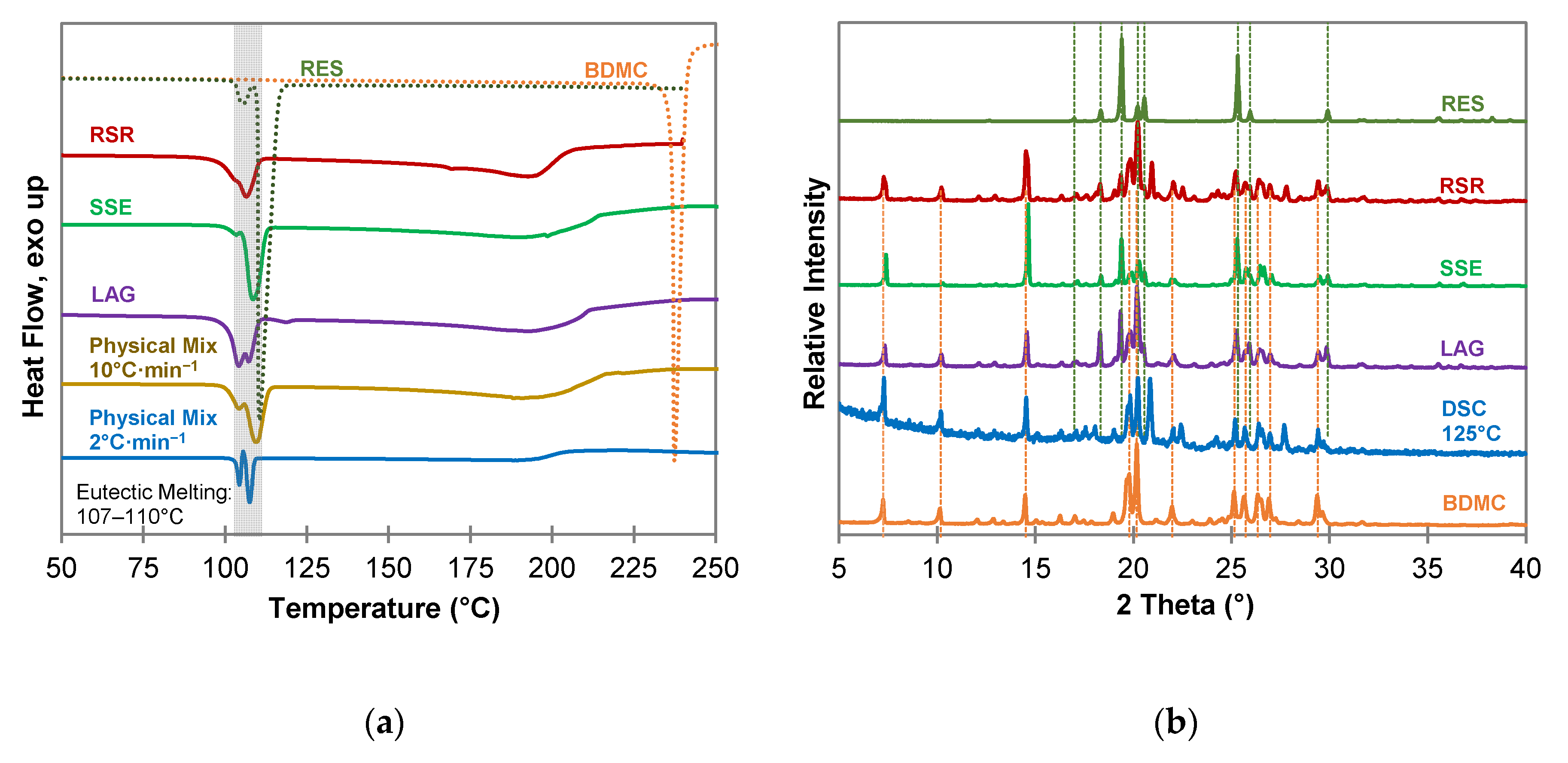
2.3. Characterization of Pyrogallol-Bis(demethoxy)curcumin Co-Crystal
2.3.1. Composition of the PYR-BDMC Co-Crystal
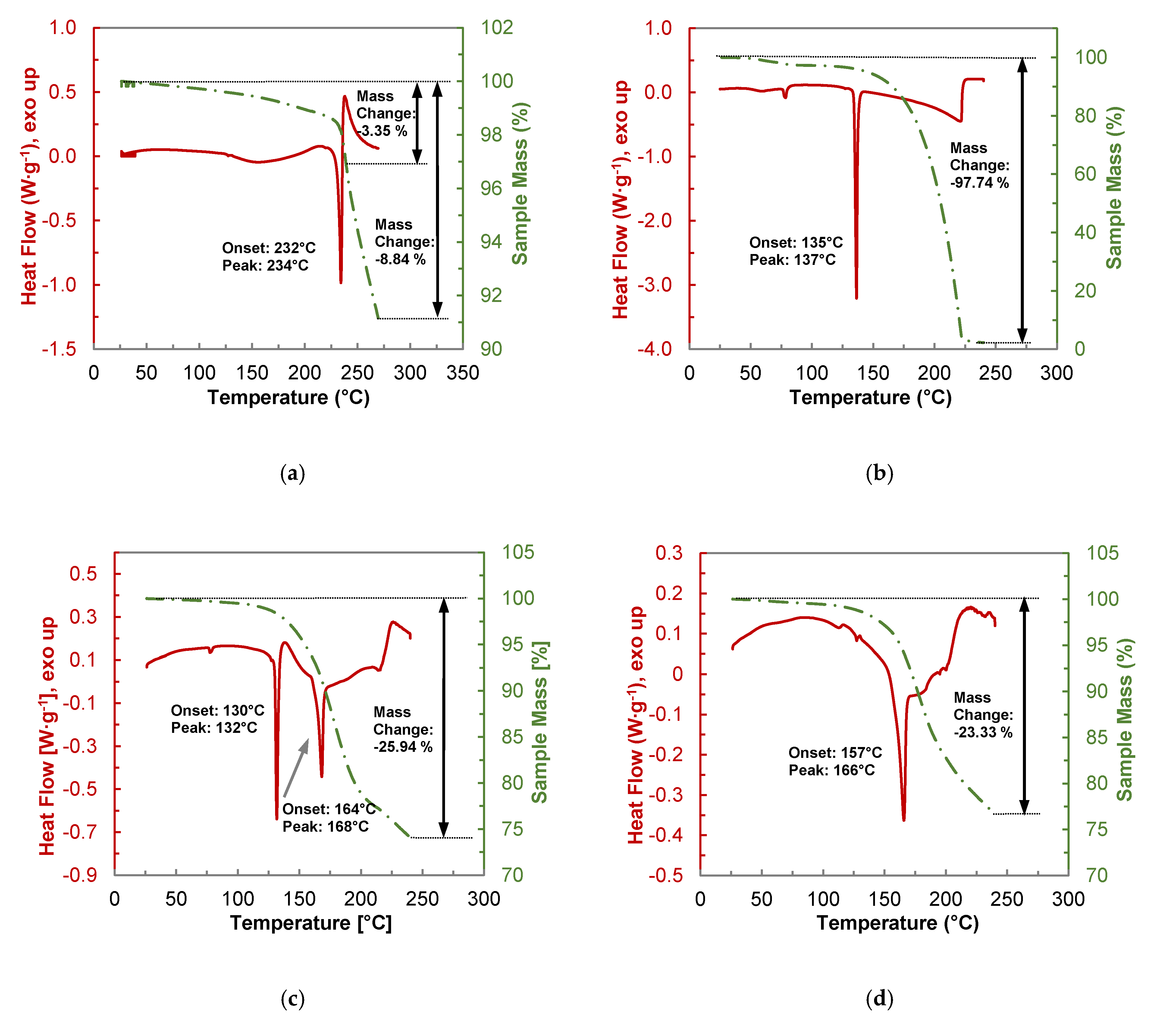
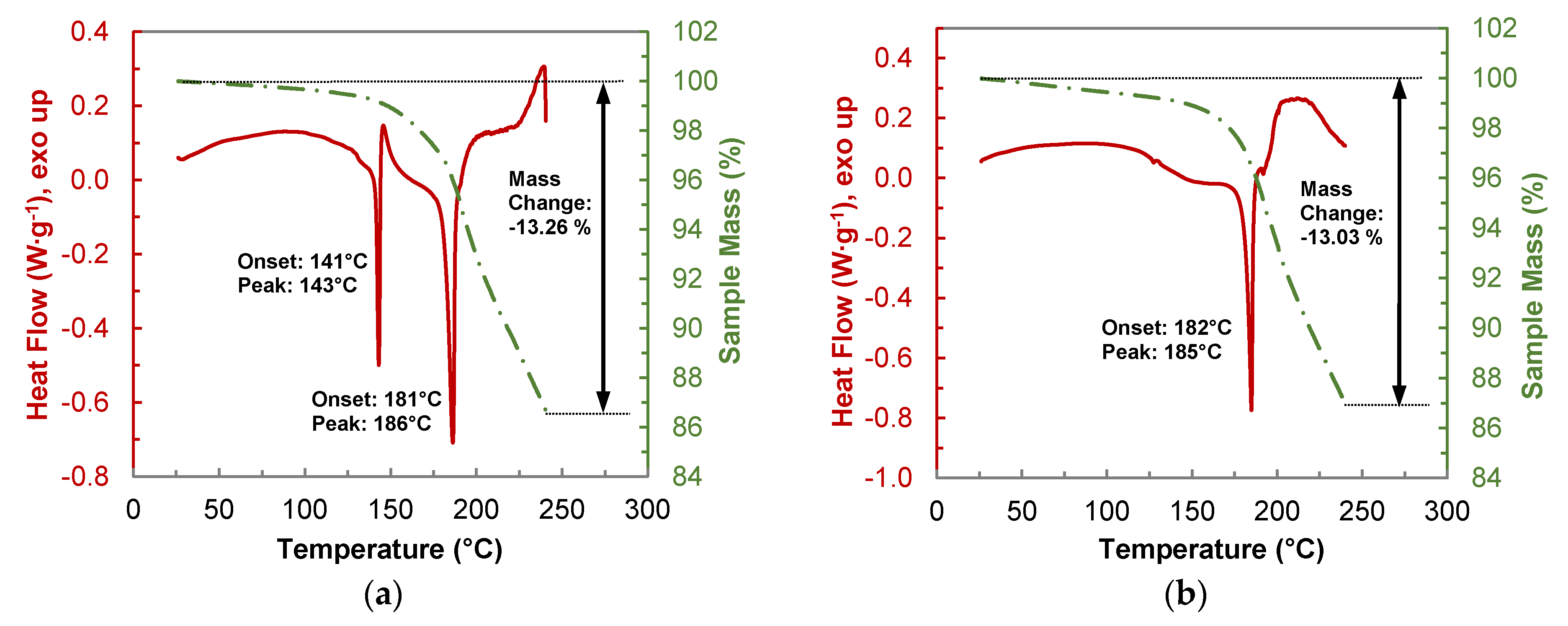
2.3.2. Thermogravimetric Analysis of the PYR-BDMC Co-Crystal and its Single Components
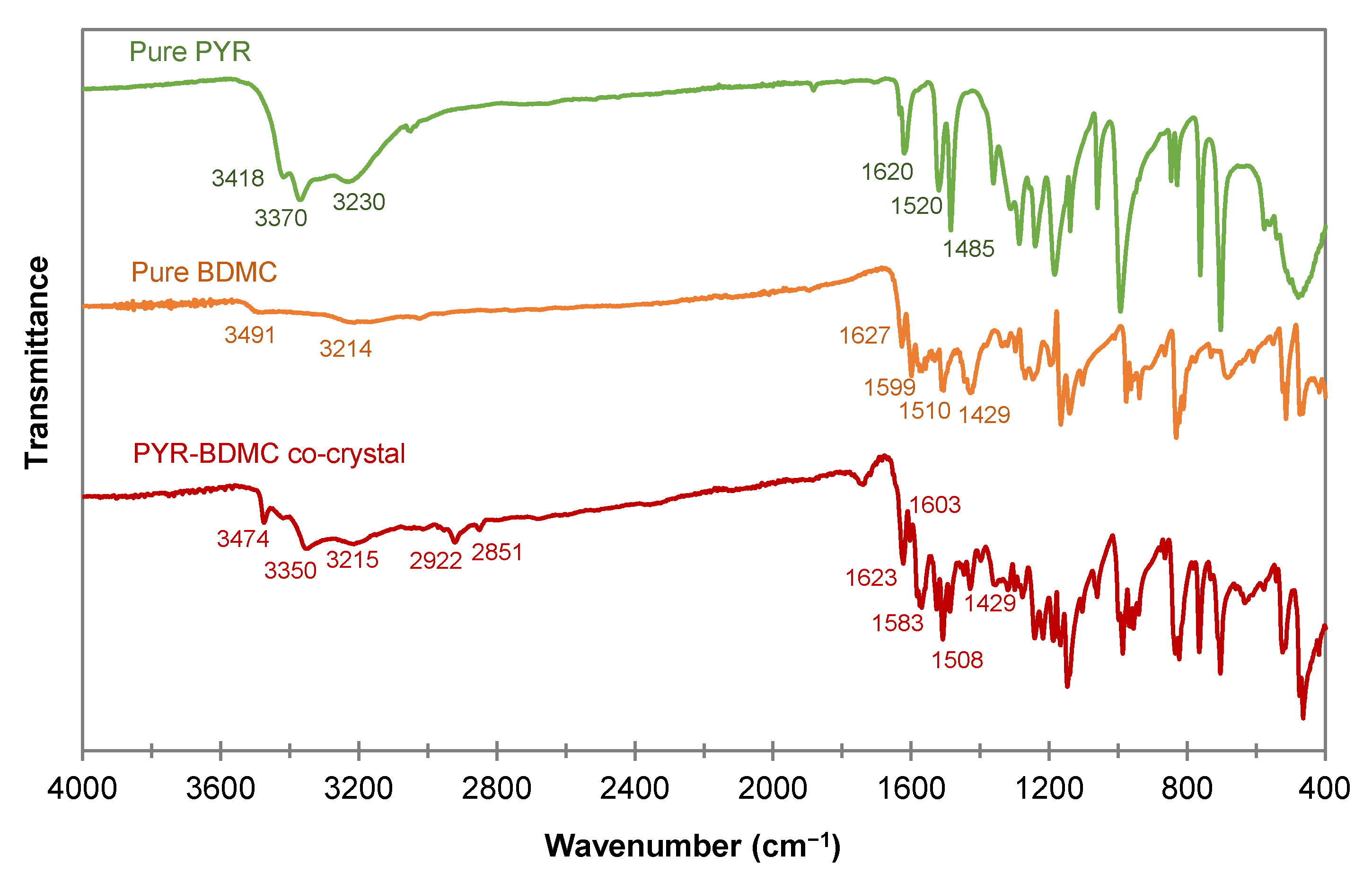
2.3.3. Fourier Transform Infrared Spectroscopy (FTIR) of the PYR-BDMC Co-Crystal and its Single Components
2.3.4. Microscopic Analysis
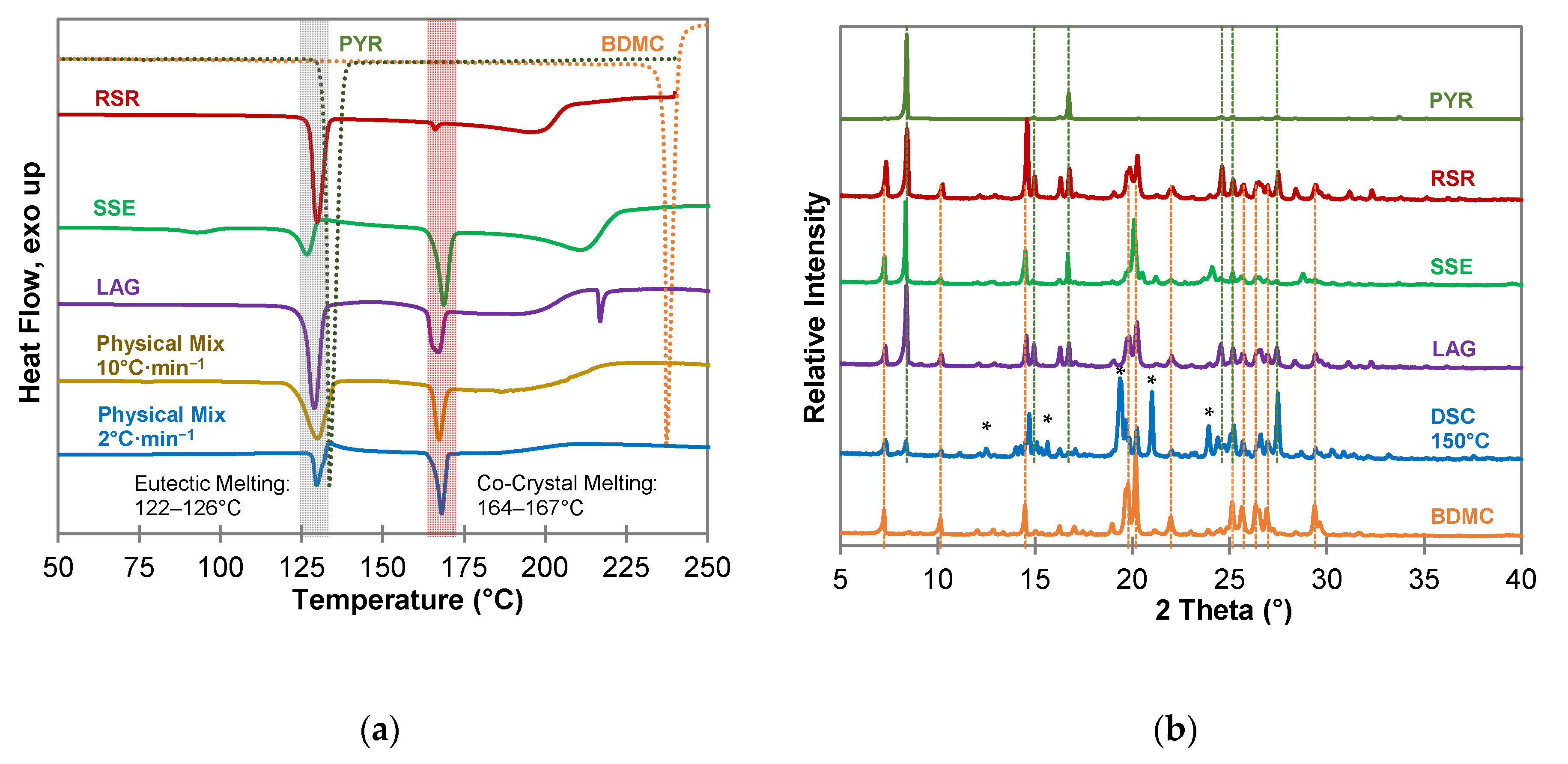
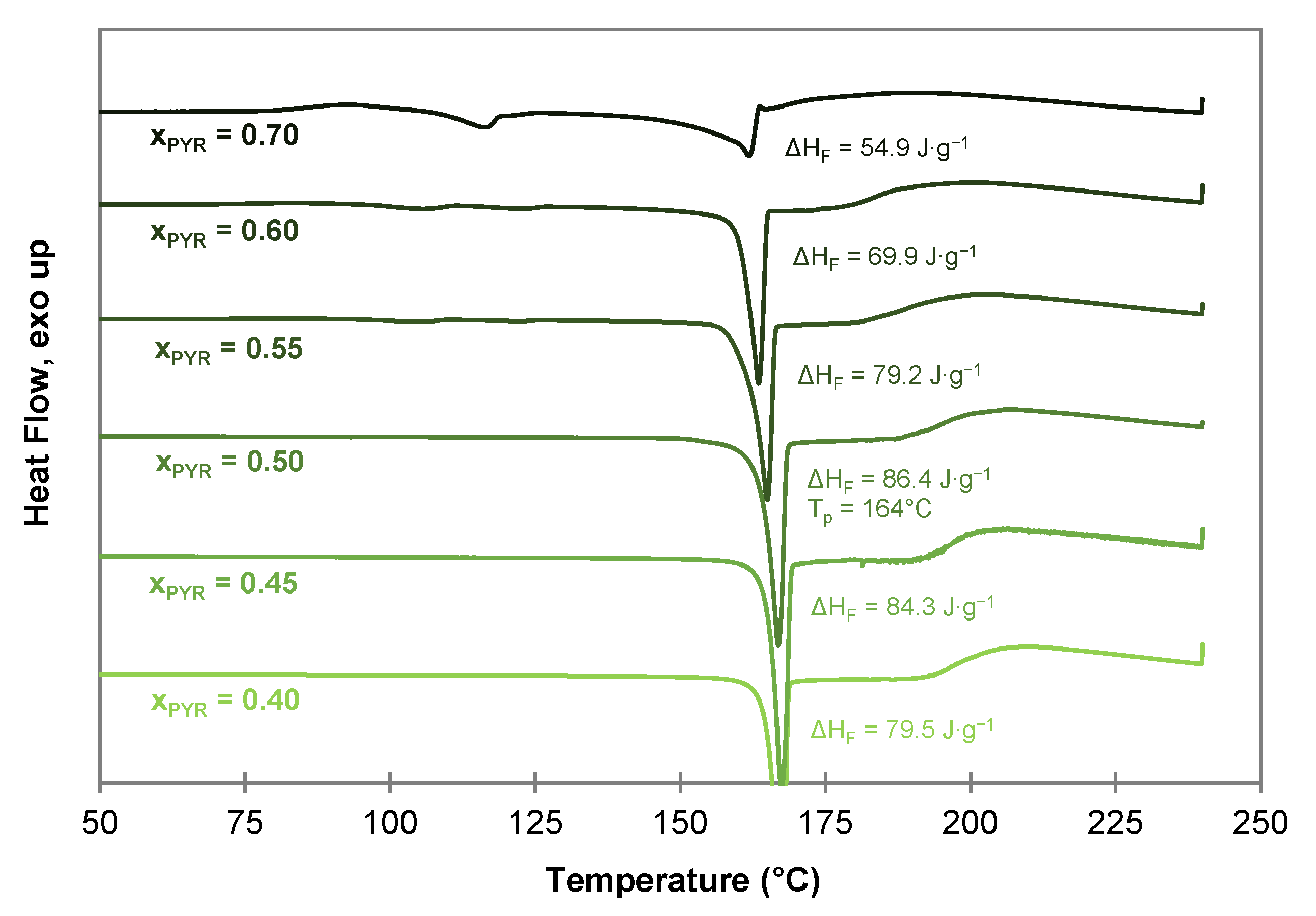
2.4. Characterization of Hydroxyquinol-Bis(demethoxy)curcumin Co-Crystal
2.4.1. Composition of the HYQ-BDMC Co-Crystal
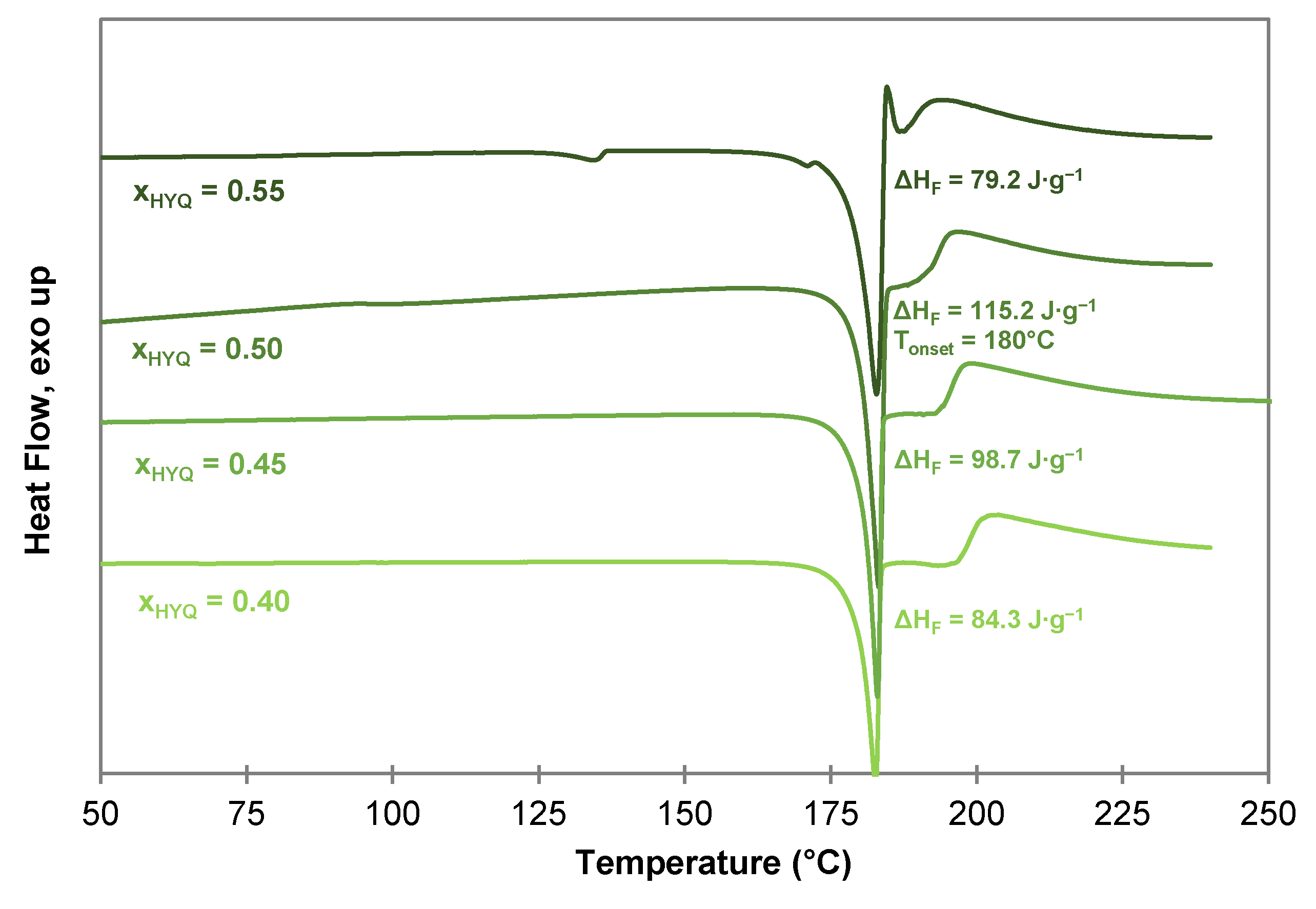
2.4.2. Thermogravimetric Analysis of the HYQ-BDMC Co-Crystal and its Single Components
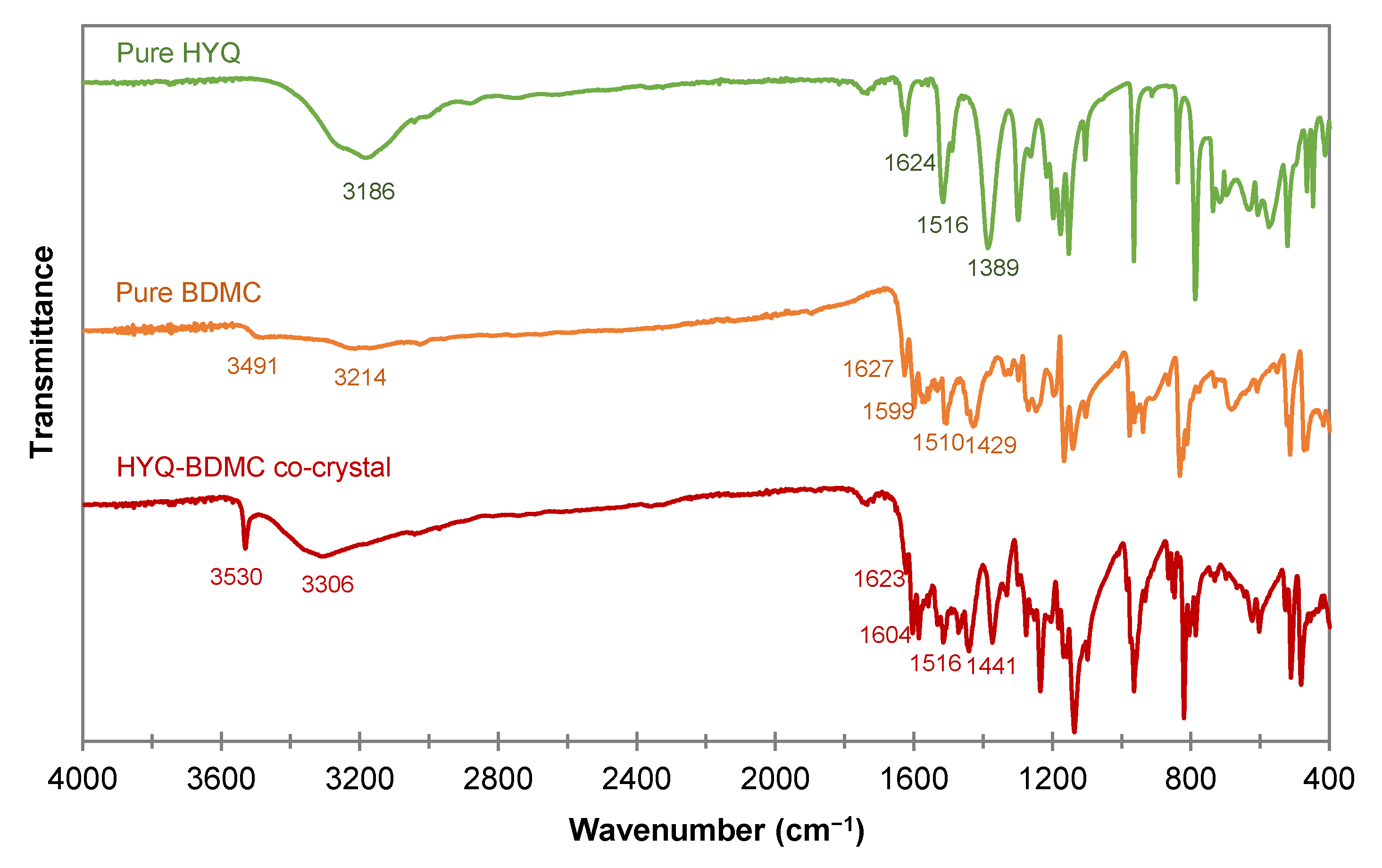
2.4.3. Fourier Transform Infrared Spectroscopy of the HYQ-BDMC Co-Crystal and its Single Components
2.4.4. Solubility Measurements
2.4.5. Microscopic Analysis
3. Materials and Methods
3.1. Materials
3.2. Preparation of BDMC Co-Crystals
3.2.1. Liquid-Assisted Grinding (LAG)
3.2.2. Crystallization from Eutectic Melt
3.2.3. Crystallization from Solution
3.3. Powder X-ray Diffraction (PXRD)
3.4. Differential Scanning Calorimetry (DSC)
3.5. Thermogravimetric Analysis (TGA)
3.6. Fourier Transform Infrared Spectroscopy (FTIR)
3.7. Solubility Measurements
3.8. Microscopic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- DerMarderosian, A.; Beutler, J.A. The review of Natural Products: The Most Complete Source of Natural Product Information, 8th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2014; ISBN 978-1-57-439368-2. [Google Scholar]
- Ahmed, T.; Gilani, A.-H. Therapeutic potential of turmeric in Alzheimer’s disease: Curcumin or curcuminoids? Phytother. Res. 2014, 28, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Ak, T.; Gülçin, I. Antioxidant and radical scavenging properties of curcumin. Chem. Biol. Interact. 2008, 174, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.M.; Teter, B.; Frautschy, S.A. Neuroprotective effects of curcumin. Adv. Exp. Med. Biol. 2007, 595, 197–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzaei, M.H.; Zobeiri, M.; Parvizi, F.; El-Senduny, F.F.; Marmouzi, I.; Coy-Barrera, E.; Naseri, R.; Nabavi, S.M.; Rahimi, R.; Abdollahi, M. Curcumin in Liver Diseases: A Systematic Review of the Cellular Mechanisms of Oxidative Stress and Clinical Perspective. Nutrients 2018, 10, 855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olotu, F.; Agoni, C.; Soremekun, O.; Soliman, M.E.S. An Update on the Pharmacological Usage of Curcumin: Has it Failed in the Drug Discovery Pipeline? Cell Biochem. Biophys. 2020, 78, 267–289. [Google Scholar] [CrossRef]
- Patel, S.S.; Acharya, A.; Ray, R.S.; Agrawal, R.; Raghuwanshi, R.; Jain, P. Cellular and molecular mechanisms of curcumin in prevention and treatment of disease. Crit. Rev. Food Sci. Nutr. 2020, 60, 887–939. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Tyagi, A.K.; Aggarwal, B.B. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: The golden pigment from golden spice. Cancer Res. Treat. 2014, 46, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.-A.; Kim, B.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Curcumin induces apoptosis by inhibiting sarco/endoplasmic reticulum Ca2+ ATPase activity in ovarian cancer cells. Cancer Lett. 2016, 371, 30–37. [Google Scholar] [CrossRef]
- Wiggers, H.J.; Zaioncz, S.; Cheleski, J.; Mainardes, R.M.; Khalil, N.M. Curcumin, a Multitarget Phytochemical: Challenges and perspectives. Stud. Nat. Prod. Chem. 2017, 53, 243–276. [Google Scholar] [CrossRef]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef]
- Basile, V.; Ferrari, E.; Lazzari, S.; Belluti, S.; Pignedoli, F.; Imbriano, C. Curcumin derivatives: Molecular basis of their anti-cancer activity. Biochem. Pharmacol. 2009, 78, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonrao, M.; Yodkeeree, S.; Ampasavate, C.; Anuchapreeda, S.; Limtrakul, P. The inhibitory effect of turmeric curcuminoids on matrix metalloproteinase-3 secretion in human invasive breast carcinoma cells. Arch. Pharm. Res. 2010, 33, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Yodkeeree, S.; Chaiwangyen, W.; Garbisa, S.; Limtrakul, P. Curcumin, demethoxycurcumin and bisdemethoxycurcumin differentially inhibit cancer cell invasion through the down-regulation of MMPs and uPA. J. Nutr. Biochem. 2009, 20, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ukrainczyk, M.; Hodnett, B.K.; Rasmuson, Å.C. Process Parameters in the Purification of Curcumin by Cooling Crystallization. Org. Process. Res. Dev. 2016, 20, 1593–1602. [Google Scholar] [CrossRef]
- Horosanskaia, E.; Yuan, L.; Seidel-Morgenstern, A.; Lorenz, H. Purification of Curcumin from Ternary Extract-Similar Mixtures of Curcuminoids in a Single Crystallization Step. Crystals 2020, 10, 206. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, C.; Ukrainczyk, M.; Gamidi, R.K.; Hodnett, B.K.; Rasmuson, Å.C. Extraction and Purification of Curcuminoids from Crude Curcumin by a Combination of Crystallization and Chromatography. Org. Process. Res. Dev. 2017, 21, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Griesser, U.J. The Importance of Solvates. In Polymorphism: In the Pharmaceutical Industry; Hilfiker, R., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 211–233. ISBN 9783527607884. [Google Scholar]
- Urbanus, J.; Roelands, C.P.M.; Verdoes, D.; Jansens, P.J.; ter Horst, J.H. Co-Crystallization as a Separation Technology: Controlling Product Concentrations by Co-Crystals. Cryst. Growth Des. 2010, 10, 1171–1179. [Google Scholar] [CrossRef]
- Chow, S.F.; Shi, L.; Ng, W.W.; Leung, K.H.Y.; Nagapudi, K.; Sun, C.C.; Chow, A.H.L. Kinetic Entrapment of a Hidden Curcumin Cocrystal with Phloroglucinol. Cryst. Growth Des. 2014, 14, 5079–5089. [Google Scholar] [CrossRef]
- Sanphui, P.; Goud, N.R.; Khandavilli, U.B.R.; Nangia, A. Fast Dissolving Curcumin Cocrystals. Cryst. Growth Des. 2011, 11, 4135–4145. [Google Scholar] [CrossRef]
- Sathisaran, I.; Dalvi, S.V. Crystal Engineering of Curcumin with Salicylic Acid and Hydroxyquinol as Coformers. Cryst. Growth Des. 2017, 17, 3974–3988. [Google Scholar] [CrossRef]
- Wong, S.N.; Hu, S.; Ng, W.W.; Xu, X.; Lai, K.L.; Lee, W.Y.T.; Chow, A.H.L.; Sun, C.C.; Chow, S.F. Cocrystallization of Curcumin with Benzenediols and Benzenetriols via Rapid Solvent Removal. Cryst. Growth Des. 2018, 18, 5534–5546. [Google Scholar] [CrossRef]
- Ribas, M.M.; Aguiar, G.P.S.; Muller, L.G.; Siebel, A.M.; Lanza, M.; Oliveira, J.V. Curcumin-nicotinamide cocrystallization with supercritical solvent (CSS): Synthesis, characterization and in vivo antinociceptive and anti-inflammatory activities. Ind. Crop. Prod. 2019, 139, 111537. [Google Scholar] [CrossRef]
- Chava, S.; Gorantla, S.R.A.; Muppidi, V.K. Solid Forms of Curcumin and Derivatives Thereof. International Patent Application No. PCT/IB2014/002034, 6 October 2014. [Google Scholar]
- Rathi, N.; Paradkar, A.; Gaikar, V.G. Polymorphs of Curcumin and Its Cocrystals with Cinnamic Acid. J. Pharm. Sci. 2019, 108, 2505–2516. [Google Scholar] [CrossRef] [Green Version]
- Pantwalawalkar, J.; More, H.; Bhange, D.; Patil, U.; Jadhav, N. Novel curcumin ascorbic acid cocrystal for improved solubility. J. Drug Deliv. Sci. Technol. 2020, 102233. [Google Scholar] [CrossRef]
- Su, H.; He, H.; Tian, Y.; Zhao, N.; Sun, F.; Zhang, X.; Jiang, Q.; Zhu, G. Syntheses and characterizations of two curcumin-based cocrystals. Inorg. Chem. Commun. 2015, 55, 92–95. [Google Scholar] [CrossRef]
- Yuan, L.; Lorenz, H. Solvate Formation of Bis(demethoxy)curcumin: Screening and Characterization. Crystals 2018, 8, 407. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Horosanskaia, E.; Engelhardt, F.; Edelmann, F.T.; Couvrat, N.; Sanselme, M.; Cartigny, Y.; Coquerel, G.; Seidel-Morgenstern, A.; Lorenz, H. Solvate Formation of Bis(demethoxy)curcumin: Crystal Structure Analyses and Stability Investigations. Cryst. Growth Des. 2019, 19, 854–867. [Google Scholar] [CrossRef]
- Leyssens, T.; Tumanova, N.; Robeyns, K.; Candoni, N.; Veesler, S. Solution cocrystallization, an effective tool to explore the variety of cocrystal systems: Caffeine/dicarboxylic acid cocrystals. CrystEngComm 2014, 16, 9603–9611. [Google Scholar] [CrossRef]
- Boldyreva, E. Mechanochemistry of inorganic and organic systems: What is similar, what is different? Chem. Soc. Rev. 2013, 42, 7719–7738. [Google Scholar] [CrossRef]
- Germann, L.S.; Arhangelskis, M.; Etter, M.; Dinnebier, R.E.; Friščić, T. Challenging the Ostwald rule of stages in mechanochemical cocrystallisation. Chem. Sci. 2020, 11, 10092–10100. [Google Scholar] [CrossRef]
- Yamashita, H.; Hirakura, Y.; Yuda, M.; Teramura, T.; Terada, K. Detection of cocrystal formation based on binary phase diagrams using thermal analysis. Pharm. Res. 2013, 30, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Hirakura, Y.; Yuda, M.; Terada, K. Coformer screening using thermal analysis based on binary phase diagrams. Pharm. Res. 2014, 31, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Springuel, G.; Norberg, B.; Robeyns, K.; Wouters, J.; Leyssens, T. Advances in Pharmaceutical Co-crystal Screening: Effective Co-crystal Screening through Structural Resemblance. Cryst. Growth Des. 2012, 12, 475–484. [Google Scholar] [CrossRef]
- Wünsche, S. Basic Studies of Co-crystal Formation of a Pharmaceutically Relevant Substance. Master’s Thesis, Otto von Guericke University, Magdeburg, Germany, 2019. [Google Scholar]
- Robertson, J.M.; Ubbelohde, A.R.; Bragg, W.H. A new form of resorcinol. II. Thermodynamic properties in relation to structure. Proc. R. Soc. A 1938, 167, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Safari, F.; Olejniczak, A.; Katrusiak, A. Pressure-Dependent Crystallization Preference of Resorcinol Polymorphs. Cryst. Growth Des. 2019, 19, 5629–5635. [Google Scholar] [CrossRef]
- Thakuria, R.; Cherukuvada, S.; Nangia, A. Crystal Structures of Pyrogallol, Its Hydrate, and Stable Multiple Z Cocrystals with N-Heterocycles Containing Metastable Conformers of Pyrogallol. Cryst. Growth Des. 2012, 12, 3944–3953. [Google Scholar] [CrossRef]
- Péret-Almeida, L.; Cherubino, A.P.F.; Alves, R.J.; Dufossé, L.; Glória, M.B.A. Separation and determination of the physico-chemical characteristics of curcumin, demethoxycurcumin and bisdemethoxycurcumin. Food Res. Int. 2005, 38, 1039–1044. [Google Scholar] [CrossRef]
- Karlsen, J.; Mostad, A.; Tønnesen, H.H.; Hörnfeldt, A.-B.; Lönnberg, H.; Berg, J.-E.; Bartók, M.; Pelczer, I.; Dombi, G. Structural Studies of Curcuminoids. VI. Crystal Structure of 1,7-Bis(4-hydroxyphenyl)-1,6-heptadiene-3,5-dione Hydrate. Acta Chem. Scand. 1988, 42b, 23–27. [Google Scholar] [CrossRef]
- Carl Roth. Safety Data Sheet: Pyrogallol; Carl Roth: Karlsruhe, Germany, 2019; No. 3963. [Google Scholar]
- Günzler, H.; Gremlich, H.-U. IR-Spektroskopie. Eine Einführung, 4th ed.; Vollständig Überarbeitete und Aktualisierte Aufl.; Wiley-VCH: Hoboken, NJ, USA, 2003; ISBN 3-527-30801-6. [Google Scholar]
- Fiege, H.; Voges, H.-W.; Hamamoto, T.; Umemura, S.; Iwata, T.; Miki, H.; Fujita, Y.; Buysch, H.-J.; Garbe, D.; Paulus, W. Phenol Derivatives. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PYR | BDMC | PYR-BDMC Co-Crystal | |
|---|---|---|---|
| Phenolic OH stretching (cm−1) | 3418, 3370, 3230 | 3491, 3214 | 3474, 3350, 3215 |
| C=O stretching (cm−1) | N.A. | 1627 | 1623 |
| Aromatic C=C stretching (cm−1) | 1620 | 1599 | 1603 |
| In-plane bending of enol C-O (cm−1) | N.A. | 1429 | 1429 |
| Methylene C-H stretching (cm−1) | N.A. | N.A. | 2922, 2851 |
| HYQ | BDMC | HYQ-BDMC Co-Crystal | |
|---|---|---|---|
| Phenolic OH stretching (cm−1) | 3186 | 3491, 3214 | 3530, 3306 |
| C=O stretching (cm−1) | N.A. | 1627 | 1623 |
| Aromatic C=C stretching (cm−1) | 1624 | 1599 | 1604 |
| In-plane bending of enol C-O (cm−1) | N.A. | 1429 | 1441 |
| csat in EtOH (g·L−1) | csat in EtOH/H2O (50/50 v/v) (g·L−1) | |
|---|---|---|
| Pure BDMC | 43.25 ± 0.58 | 0.38 ± 0.05 |
| BDMC within co-crystal | 39.04 ± 1.10 | 0.83 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wünsche, S.; Yuan, L.; Seidel-Morgenstern, A.; Lorenz, H. A Contribution to the Solid State Forms of Bis(demethoxy)curcumin: Co-Crystal Screening and Characterization. Molecules 2021, 26, 720. https://doi.org/10.3390/molecules26030720
Wünsche S, Yuan L, Seidel-Morgenstern A, Lorenz H. A Contribution to the Solid State Forms of Bis(demethoxy)curcumin: Co-Crystal Screening and Characterization. Molecules. 2021; 26(3):720. https://doi.org/10.3390/molecules26030720
Chicago/Turabian StyleWünsche, Steffi, Lina Yuan, Andreas Seidel-Morgenstern, and Heike Lorenz. 2021. "A Contribution to the Solid State Forms of Bis(demethoxy)curcumin: Co-Crystal Screening and Characterization" Molecules 26, no. 3: 720. https://doi.org/10.3390/molecules26030720
APA StyleWünsche, S., Yuan, L., Seidel-Morgenstern, A., & Lorenz, H. (2021). A Contribution to the Solid State Forms of Bis(demethoxy)curcumin: Co-Crystal Screening and Characterization. Molecules, 26(3), 720. https://doi.org/10.3390/molecules26030720