
Spectroscopic and Theoretical Study of the Intramolecular π-Type Hydrogen Bonding and Conformations of 2-Cyclopenten-1-ol


Abstract
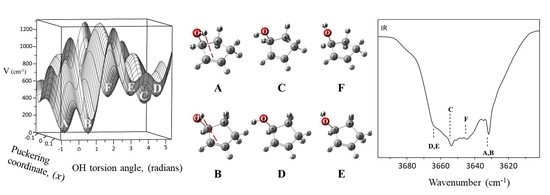
:
1. Introduction
2. Results and Discussion
2.1. Calculated Molecular Conformations
2.2. Vibrational Spectra
2.3. Potential Energy Surface
3. Materials and Methods
3.1. Computations
3.1.1. Structure and Frequency Calculations
3.1.2. Potential Energy Surface
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Laane, J.; Ocola, E.J.; Chun, H.J. Vibrational Potential Energy Surfaces in Ground and Electronic Excited States. In Frontiers and Advances in Molecular Spectroscopy; Laane, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 101–142. [Google Scholar] [CrossRef]
- Laane, J. Vibrational Potential Energy Surfaces in Electronic Excited States. In Frontiers of Molecular Spectroscopy; Laane, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 63–132. [Google Scholar] [CrossRef]
- Laane, J. Experimental Determination of Vibrational Potential Energy Surfaces and Molecular Structures in Electronic Excited States. J. Chem. Phys. A 2000, 104, 7715–7733. [Google Scholar] [CrossRef]
- Laane, J. Spectroscopic Determination of Ground and Excited State Vibrational Potential Energy Surfaces. Int. Rev. Phys. Chem. 1999, 18, 301–341. [Google Scholar] [CrossRef]
- Laane, J. Vibrational Potential Energy Surfaces and Conformations of Molecules in Ground and Excited Electronic States. Annu. Rev. Phys. Chem. 1994, 45, 179–211. [Google Scholar] [CrossRef]
- Laane, J. Vibrational Potential Energy Surfaces of Non-Rigid Molecules in Ground and Excited Electronic States. In Structures and Conformations of Non-Rigid Molecules; Laane, J., Dakkouri, M., Eds.; Kluwer Publishing: Amsterdam, The Netherlands, 1993; pp. 65–98. [Google Scholar] [CrossRef]
- Laane, J. Determination of Vibrational Potential Energy Surfaces from Raman and Infrared Spectra. J. Pure Appl. Chem. 1987, 59, 1307–1326. [Google Scholar] [CrossRef] [Green Version]
- Al-Saadi, A.A.; Wagner, M.; Laane, J. Spectroscopic and Computational Studies of the Intramolecular Hydrogen Bonding of 2-Indanol. J. Phys. Chem. A 2006, 110, 12292–12297. [Google Scholar] [CrossRef]
- Ocola, E.J.; Al-Saadi, A.A.; Mlynek, C.; Hopf, H.; Laane, J. Intramolecular π-Type Hydrogen Bonding and Conformations of 3-Cyclopenten-1-ol. 2. Infrared and Raman Spectral Studies at High Temperatures. J. Phys. Chem. A 2010, 114, 7457–7461. [Google Scholar] [CrossRef]
- Al-Saadi, A.A.; Ocola, E.J.; Laane, J. Intramolecular π-Type Hydrogen Bonding and Conformations of 3-Cyclopenten-1-ol. 1. Theoretical Calculations. J. Phys. Chem. A 2010, 114, 7453–7456. [Google Scholar] [CrossRef] [PubMed]
- Ocola, E.J.; Laane, J. Spectroscopic and Theoretical Study of the Intramolecular π-Type Hydrogen Bonding and Conformations of 2-Cyclohexen-1-ol. J. Phys Chem. A 2016, 120, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ocola, E.J.; Laane, J. Theoretical Investigation of Intramolecular π-Type Hydrogen Bonding and Internal Rotation of 2-Cyclopropen-1-ol, 2-Cyclopropen-1-thiol and 2-Cyclopropen-1-amine. Mol. Phys. 2019, 17, 1404–1412. [Google Scholar] [CrossRef]
- Ocola, E.J.; Laane, J. Spectroscopic and Theoretical Study of the Intramolecular π-type Hydrogen Bonding and Conformations of 3-Cyclopentene-1-amine. J. Phys. Chem. 2020, 124, 5907–5916. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.W.; Shulgin, A.T. Intramolecular Hydrogen Bonds to π-Electrons and Other Weakly Basic Groups. J. Am. Chem. Soc. 1958, 80, 5358–5363. [Google Scholar] [CrossRef]
- Smith, Z.; Carballo, N.; Wilson, E.B.; Marstokk, K.-M.; Møllendal, H. Conformations, Possible H Bonding, and Microwave Spectrum of 3-Buten-2-ol. J. Am. Chem. Soc. 1951, 107, 1951–1957. [Google Scholar] [CrossRef]
- Murty, A.N.; Curl, R.F., Jr. Microwave Spectrum of Allyl Alcohol. J. Chem. Phys. 1967, 46, 4176–4180. [Google Scholar] [CrossRef]
- Marstokk, K.-M.; Møllendal, H. Microwave Spectrum, Conformation and Intramolecular Hydrogen Bonding of 1,4-Pentadien-3-ol. Acta Chem. Scand. 1990, 44, 18–22. [Google Scholar] [CrossRef]
- Bräse, S.; Klæboe, P.; Marstokk, K.-M.; de Meijere, A.; Møllendal, H.; Nielsen, C.J. Conformational Properties of 2-Cyclopropylideneethanol as Studied by Microwave, Infrared and Raman Spectroscopy and by Ab Initio Computations. Acta Chem. Scand. 1998, 52, 1122–1136. [Google Scholar] [CrossRef] [Green Version]
- Bakke, J.M.; Bjerkeseth, L.H. The Conformational Composition of 3-Buten-1-ol, the Importance of Intramolecular Hydrogen Bonding. J. Mol. Struct. 1998, 470, 247–263. [Google Scholar] [CrossRef]
- Leonov, A.; Marstokk, K.-M.; de Meijere, A.; Møllendal, H. Microwave Spectrum, Conformational Equilibrium, Intramolecular Hydrogen Bonding, Tunneling, and Quantum Chemical Calculations for 1-Ethenylcyclopropan-1-ol. J. Phys. Chem. A 2000, 104, 4421–4428. [Google Scholar] [CrossRef]
- Rademacher, P.; Khelashvili, L.; Kowski, K. Spectroscopic and Theoretical Studies on Intramolecular OH–π Hydrogen Bonding in 4-Substituted 2-Allylphenols. Org. Biomol. Chem. 2005, 3, 2620–2625. [Google Scholar] [CrossRef]
- Miller, B.J.; Lane, J.R.; Kjaergaard, H.G. Intramolecular OH﮲﮲﮲π Interactions in Alkenols and Alkynols. Phys. Chem. Chem. Phys. 2011, 13, 14183–14193. [Google Scholar] [CrossRef]
- Mackeprang, K.; Schrøder, S.D.; Kjaergaard, H.G. Weak Intramolecular OH–π Hydrogen Bonding in Methallyl- and Allyl-Carbinol. Chem. Phys. Lett. 2013, 582, 31–37. [Google Scholar] [CrossRef]
- Al-Saadi, A.A.; Laane, J. Ab Initio and DFT Calculations for the Structure and Vibrational Spectra of Cyclopentene and its Isotopomers. J. Mol. Struct. 2007, 830, 46–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView 6.1.1, Graphical Interface; Semichem Inc.: Shawnee, KS, USA, 2000–2019. [Google Scholar]
- Waterloo Maple. Maple 2015; Waterloo Maple Inc.: Waterloo, ON, Canada, 2015. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  |  |  |  |  |  |
| 2CPOL Conformers | ||||||
| A | B | C | D | E | F | |
| Energy | ||||||
| Energy (cm−1) | 0 | 9 | 293 | 304 | 308 | 361 |
| Energy (kJ/mol) | 0 | 0.11 | 3.51 | 3.64 | 3.68 | 4.32 |
| Energy (kcal/mol) | 0 | 0.03 | 0.84 | 0.87 | 0.88 | 1.03 |
| Relative population (%) | ||||||
| At 25° | 35% | 34% | 9% | 8% | 8% | 6% |
| Angles (degrees) | ||||||
| Ring-puckering angle | −21.1° | 22.5° | −23.1° | 22.8° | 23.7° | −22.8° |
| OH internal rotation angle, ϕ | 24.8° | 39.2° | −75.7° | −91.0° | 172.3° | 166.9° |
| Distance (Å) | ||||||
| O−H14 | 0.960 | 0.960 | 0.958 | 0.958 | 0.958 | 0.958 |
| dH14 a | 2.682 | 3.001 | 3.019 | 3.335 | 3.680 | 3.554 |
| Molecule | H﮲﮲﮲π (C=C) Distance, Å | Method | Reference |
|---|---|---|---|
| 2-indanol (I) | 2.650 | CCSD/cc-pVTZ | This work |
| 2.580 | MP2/cc-pVTZ | This work | |
| 3-cyclopenten-1-ol (II) | 2.744 | CCSD/cc-pVTZ | This work |
| 2.673 | MP2/cc-pVTZ | This work | |
| 2-cyclohexen-1-ol (III) | 2.756 | CCSD/cc-pVTZ | This work |
| 2.737 | MP2/cc-pVTZ | [11] | |
| 2-cyclopropen-1-ol (IV) | 2.488 | CCSD/cc-pVTZ | [12] |
| 2.478 | MP2/cc-pVTZ | [12] | |
| 2-cyclopropen-1-thiol (V) | 2.774 | CCSD/cc-pVTZ | [12] |
| 2.740 | MP2/cc-pVTZ | [12] | |
| 2-cyclopropen-1-amine (VI) | 2.583 | CCSD/cc-pVTZ | [12] |
| 2.571 | MP2/cc-pVTZ | [12] | |
| 3-cyclopenten-1-amine (VII) | 2.850 | CCSD/cc-pVTZ | [13] |
| 2.773 | MP2/cc-pVTZ | This work | |
| 2-cyclopenten-1-ol (2CPOL) | 2.682 | CCSD/cc-pVTZ | This work |
| 2.632 | MP2/cc-pVTZ | This work |
| 2-Cyclopenten-1-ol | 3-Cyclopenten-1-ol | 3-Cyclopenten-1-amine | Cyclopentene | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Observed | Calculated | Observed | |||||||||
| A | A | A | A | Puckered, Cs | |||||||
| Ring-Pseudo C2v | Freq. | IR a | Freq. | (IR, R) b | Freq. c | (IR, R) d,e | Freq. f | (IR, R) d,f | Freq. e | (IR, R) b,e | |
| A1 | |||||||||||
| v3 | C=C stretch | 1609 | m | 1609 | (3, 80) | 1607 | (2, 46) | 1613 | (m, 88) | 1623 | (m, 91) |
| v4 | CH2 deformation | 1500 | s | 1459 | (8, 67) | 1446f | (3, 19) | 1452 | (m, 17) | 1471f | (vw, 16) |
| v6 | =C-H in plane wag | 1077 | m | 1095 | (10, 100) | 1108f | (4, 95) | 1109 | (w, 58) | 1101 | (w, 66) |
| v8 | Ring stretch | 894 | m | 869 | (13, 60) | 832 | (5, 3) | 804 | (s, 43) | 900 | (m, 100) |
| v9 | Ring angle bend | 694 | s | 692 | (6, 9) | 745 | (51, 77) | 735 | (w, 9) | 796g | (vw, 1) |
| B1 | |||||||||||
| v17 | CH2 deformation | 1463 | m | 1436 | (7, 77) | 1446f | (17, 19) | 1437 | (m, 4) | 1445f | (m, 23) |
| B2 | |||||||||||
| v25 | CH2 rock | 924 | m | 971 | (14, 36) | 948f | (29, 17) | 934 | (m, 4) | 1047 | (s, 1) |
| v26 | =C-H out-of-plane wag | 726 | s | 719 | (20, 15) | 674 | (97, 12) | 671 | (s, 11) | 695 | (s, 1) |
| v27 | Ring puckering | --- | --- | 119 | (2, 11) | --- | (---, ---) | --- | (---, ---) | --- | (---, ---) |
| Other vibrations | |||||||||||
| OH stretch | 3632 | w | 3632 | (32, 26) | 3623 | (41, 9) | N. A. | N. A. | N. A. | N. A. | |
| C-H wag (up and down) | 1385 | m | 1386 | (80, 53) | 1395 | (38, 13) | 1379 | (m, 7) | N. A. | N. A. | |
| COH wag | 1212 | w | 1224 | (13, 14) | 1275 | (9, 3) | N. A. | N. A. | N. A. | N. A. | |
| C-O wag (up and down) | 462 | s | 487 | (5, 21) | --- | --- | N. A. | N. A. | N. A. | N. A. | |
| C-O wag (sideways) | 346 (L) | w | 388 | (12, 7) | 444 | (0.5, 2) | N. A. | N. A. | N. A. | N. A. | |
| vCO | C-O stretch | 1032 | m | 1045 | (75, 21) | 1048 | (54, 0.7) | N. A. | N. A. | N. A. | N. A. |
| OH torsion | 217 (L) | m | 313 | (100, 21) | 397h | (---, 8) | N. A. | N. A. | N. A. | N. A. | |
| Conformer | Observed | Calculated | ||
|---|---|---|---|---|
| Frequency | Shift | Frequency | Shift | |
| A | 3632 | 0 | 3632 | 0 |
| B | 3632 | 0 | 3634 | 2 |
| C | 3654 | 22 | 3648 | 16 |
| D | 3664 | 32 | 3658 | 26 |
| E | 3664 | 32 | 3656 | 24 |
| F | 3644 | 12 | 3644 | 12 |
| 2CPOL Conformers | ||||||
|---|---|---|---|---|---|---|
| A | B | C | D | E | F | |
| Energy, (cm−1) | ||||||
| CCSD/cc-pVTZ | 0 | 9 | 293 | 304 | 308 | 361 |
| MP2/cc-pVTZ | 0 | 89 | 308 | 406 | 406 | 409 |
| Ring-puckering coordinate, (Å) | ||||||
| CCSD/cc-pVTZ | −0.102 | 0.109 | −0.112 | 0.110 | 0.115 | −0.110 |
| MP2/cc-pVTZ | −0.114 | 0.118 | −0.124 | 0.118 | 0.124 | −0.122 |
| Ring-puckering angle, (degrees) | ||||||
| CCSD/cc-pVTZ | −21.1° | 22.5° | −23.1° | 22.8° | 23.7° | −22.8° |
| MP2/cc-pVTZ | −23.6° | 24.3° | −25.7° | 24.4° | 25.6° | −25.3° |
| OH internal rotation angle, (degrees) | ||||||
| CCSD/cc-pVTZ | 24.8° | 39.2° | 284.3° | 269.0° | 172.3° | 166.9° |
| MP2/cc-pVTZ | 22.6° | 38.7° | 286.4° | 268.6° | 172.3° | 166.6° |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ocola, E.J.; Laane, J. Spectroscopic and Theoretical Study of the Intramolecular π-Type Hydrogen Bonding and Conformations of 2-Cyclopenten-1-ol. Molecules 2021, 26, 1106. https://doi.org/10.3390/molecules26041106
Ocola EJ, Laane J. Spectroscopic and Theoretical Study of the Intramolecular π-Type Hydrogen Bonding and Conformations of 2-Cyclopenten-1-ol. Molecules. 2021; 26(4):1106. https://doi.org/10.3390/molecules26041106
Chicago/Turabian StyleOcola, Esther J., and Jaan Laane. 2021. "Spectroscopic and Theoretical Study of the Intramolecular π-Type Hydrogen Bonding and Conformations of 2-Cyclopenten-1-ol" Molecules 26, no. 4: 1106. https://doi.org/10.3390/molecules26041106
APA StyleOcola, E. J., & Laane, J. (2021). Spectroscopic and Theoretical Study of the Intramolecular π-Type Hydrogen Bonding and Conformations of 2-Cyclopenten-1-ol. Molecules, 26(4), 1106. https://doi.org/10.3390/molecules26041106