
Design, Synthesis, and In Silico Multitarget Pharmacological Simulations of Acid Bioisosteres with a Validated In Vivo Antihyperglycemic Effect


 , ,
, , 
Abstract
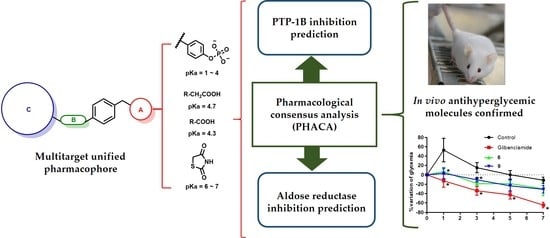
:
1. Introduction
2. Results and Discussion
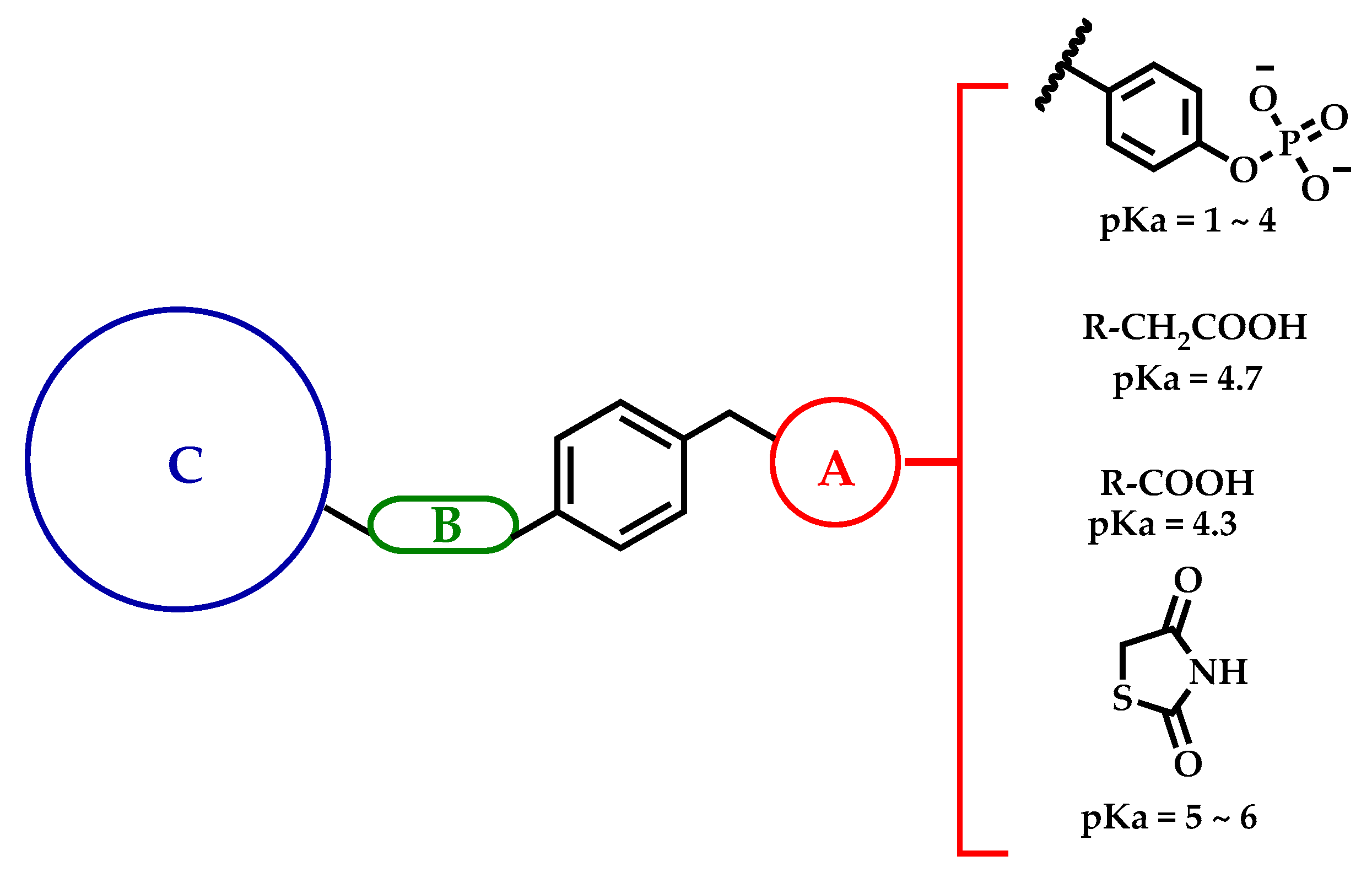
2.1. Drug Design
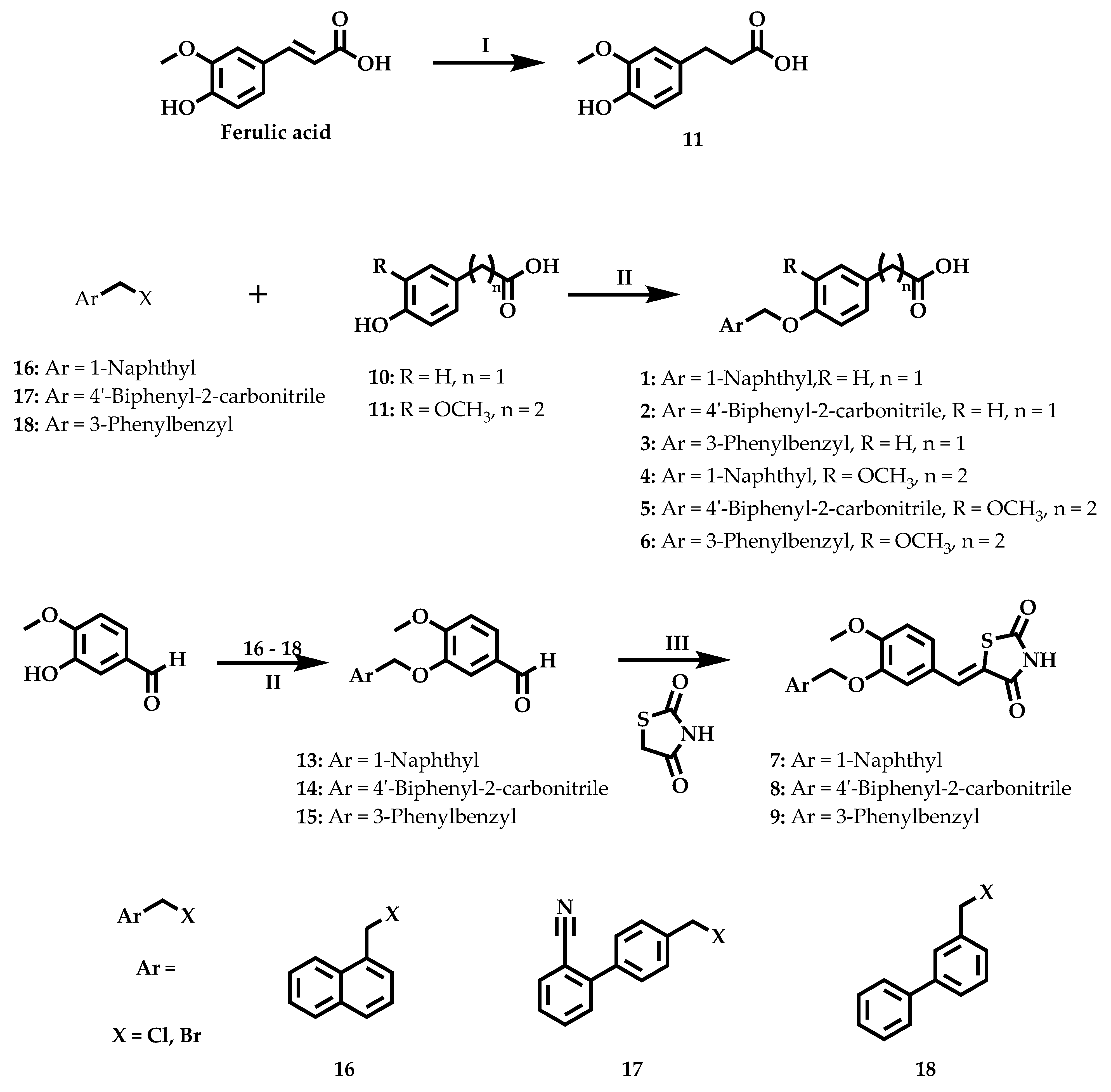
2.2. Chemistry
2.3. In Silico Analysis
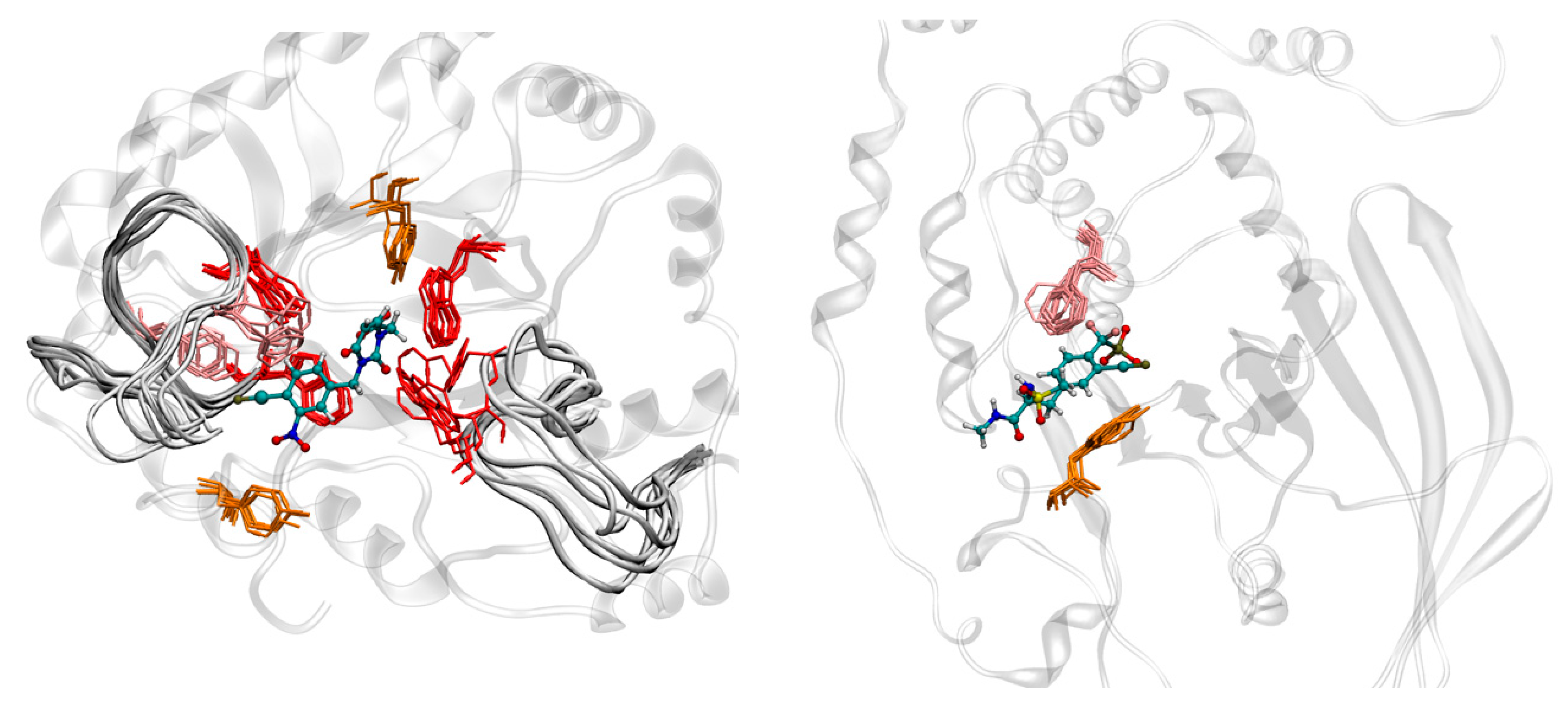
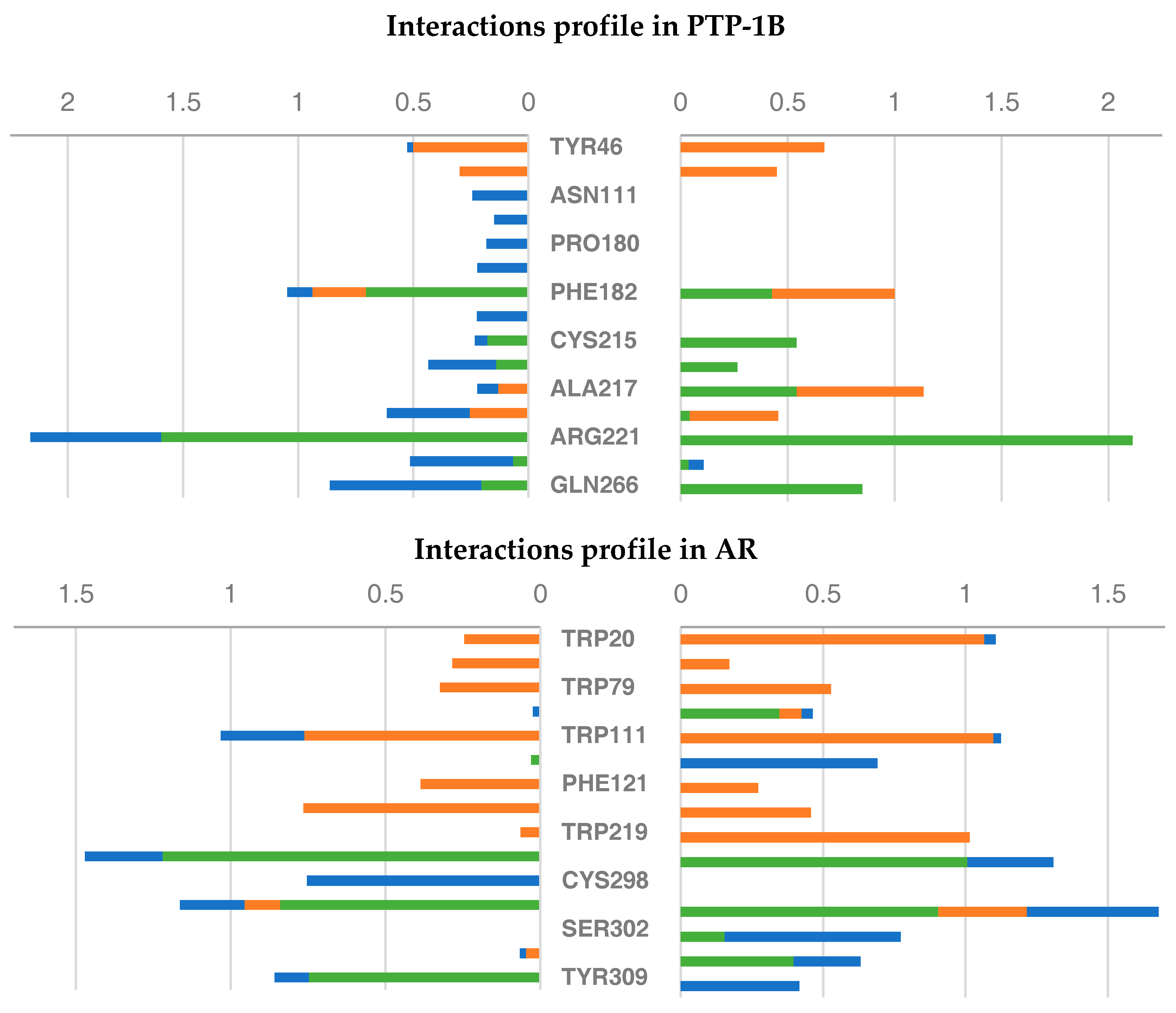
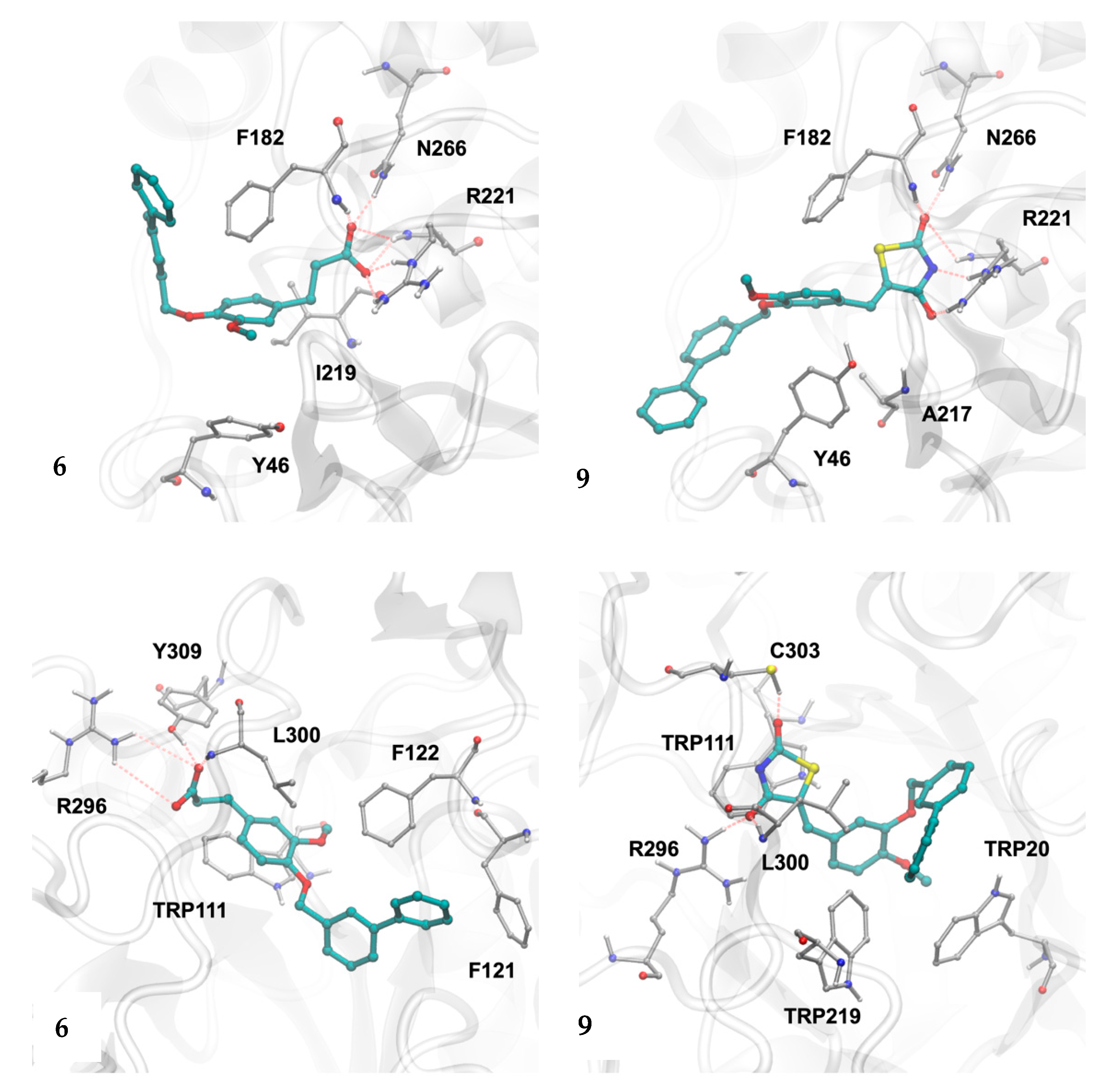
2.3.1. Structural Analysis of the Targets
2.3.2. Pharmacodynamics Predictions
2.3.3. Pharmacokinetics and Toxicological Properties
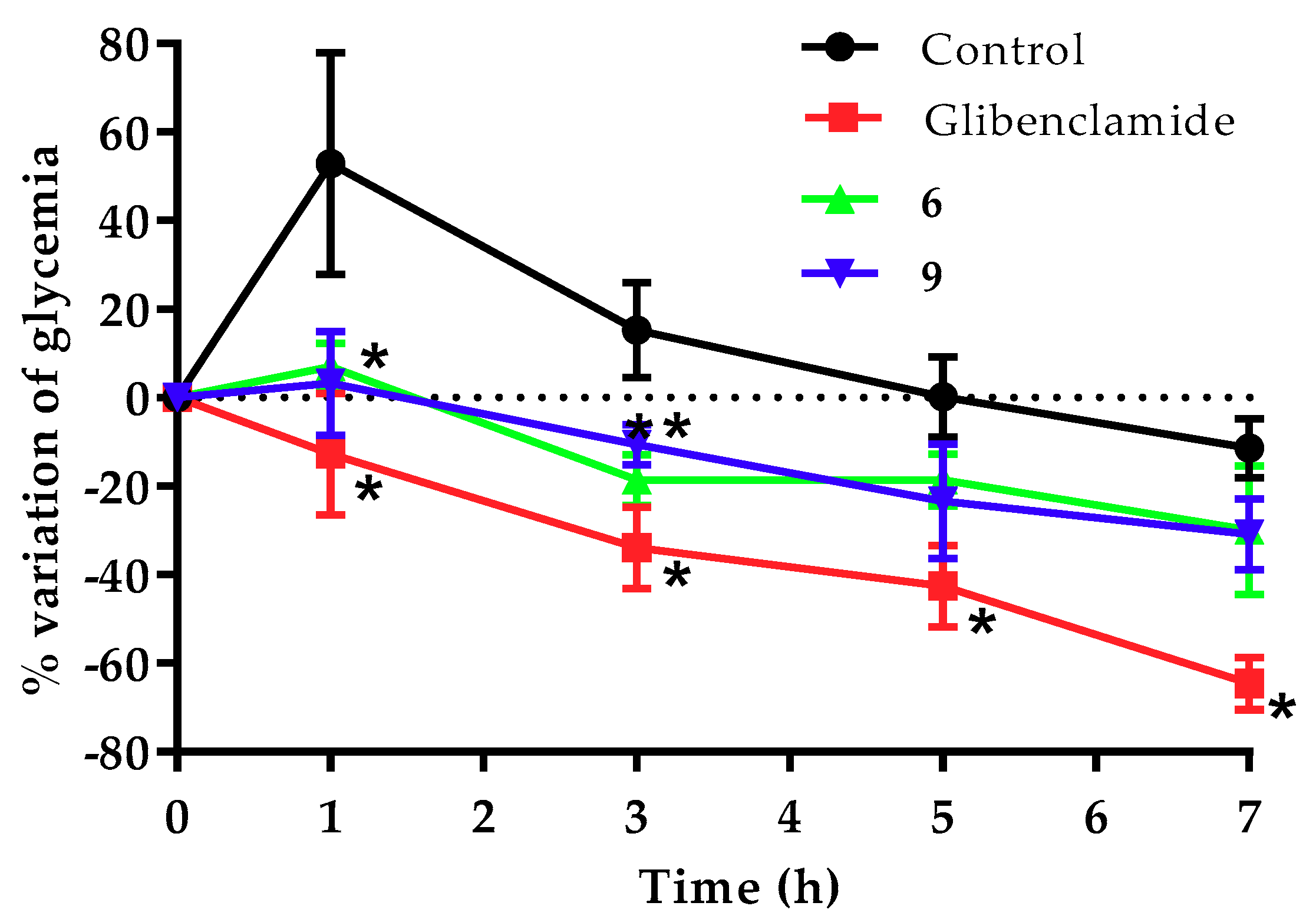
2.4. Antidiabetic Assay
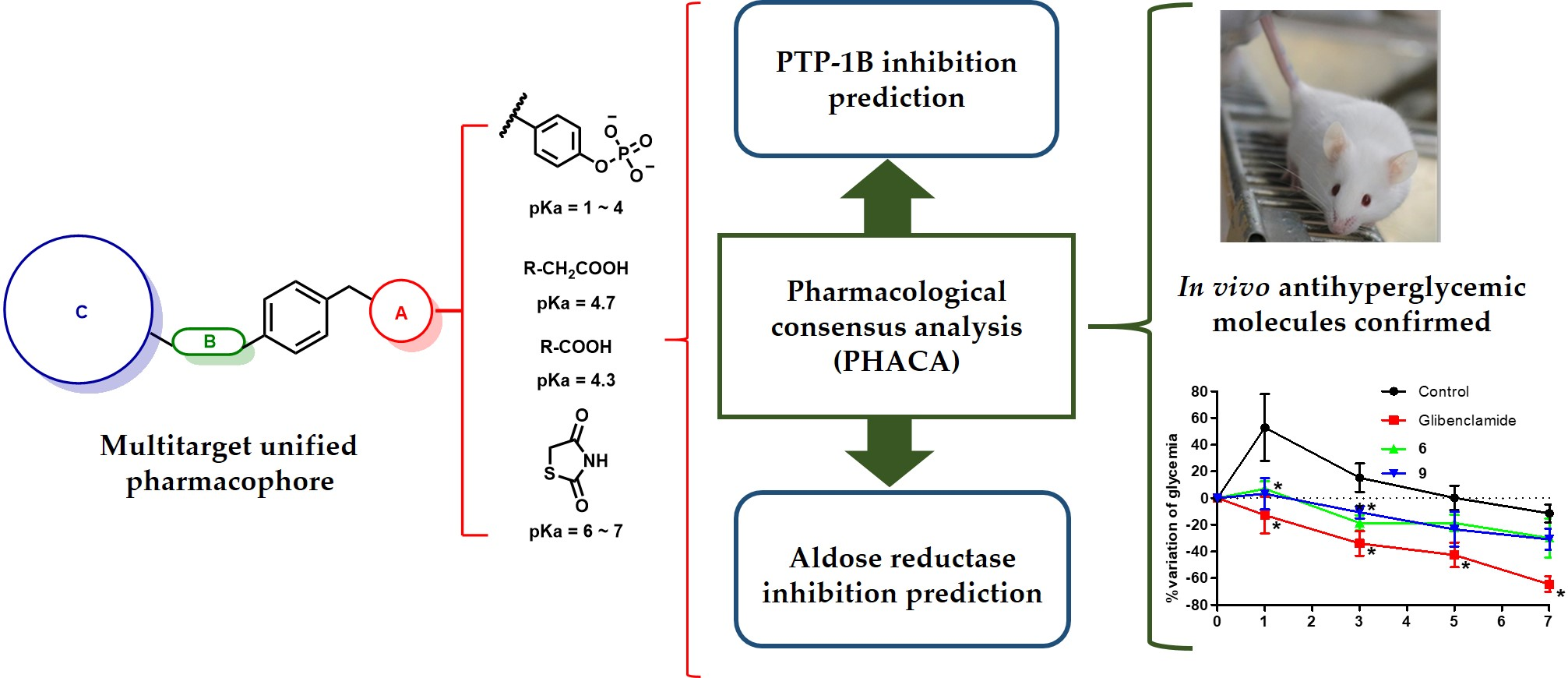
2.5. Pharmacological Consensus Analysis
2.6. Molecular Dynamics Studies of Compounds 6 and 9
3. Materials and Methods
3.1. Chemistry
3.1.1. Procedure for the Synthesis of 3-(4-hydroxy-3-methoxyphenyl)propanoic acid (11)
3.1.2. General Procedure for the Synthesis of Compounds 1–6
- [4-(1-naphthylmethoxy)phenyl]acetic acid (1)
- {4-[(2′-cyanobiphenyl-4-yl)methoxy]phenyl}acetic acid (2)
- [4-(biphenyl-3-ylmethoxy)phenyl]acetic acid (3)
- 3-[3-methoxy-4-(1-naphthylmethoxy)phenyl]propanoic acid (4)
- 3-{4-[(2′-cyanobiphenyl-4-yl)methoxy]-3-methoxyphenyl}propanoic acid (5)
- 3-[4-(biphenyl-3-ylmethoxy)-3-methoxyphenyl]propanoic acid (6)
3.1.3. General Procedure for the Synthesis of Compounds 13–15
- 4-methoxy-3-(1-naphthylmethoxy)benzaldehyde (13)
- 4′−[(5-formyl-2-methoxyphenoxy)methyl]biphenyl-2-carbonitrile (14)
- 3-(biphenyl-3-ylmethoxy)-4-methoxybenzaldehyde (15)
3.1.4. General Procedure for the Synthesis of Compounds 7–9
- (5 Z)-5-[4-methoxy-3-(1-naphthylmethoxy)benzylidene]-1,3-thiazolidine-2,4-dione (7)
- 4′-({5-[(Z)-(2,4-dioxo-1,3-thiazolidin-5-ylidene)methyl]-2-methoxyphenoxy}methyl) biphenyl-2-carbonitrile (8)
- (5 Z)-5-[3-(biphenyl-3-ylmethoxy)-4-methoxybenzylidene]-1,3-thiazolidine-2,4-dione (9)
3.2. Animals
3.2.1. Induction of Diabetes
3.2.2. Acute Antidiabetic Assay
3.3. Crystal Structures
3.4. Molecular Dynamics Simulations
3.5. Ensemble Docking
3.6. Pharmacological Consensus Analysis
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- International Diabetes Federation. IDF Diabetes Atlas 2019, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Lemanowicz, A.; Leszczyński, W.; Rusak, G.; Bialecki, M.; Ratajczak, P. Chest Adipose Tissue Distribution in Patients with Morbid Obesity. Pol. J. Radiol. 2018, 83, e68–e75. [Google Scholar] [CrossRef] [PubMed]
- Safavi, M.; Forolumadi, A.; Abdollahi, M. The Importance of Synthetic Drugs for Type 2 Diabetes Drug Discovery. Expert Opin. Drug Discov. 2013, 8, 1339–1363. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferranini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of Hyperglycemia in Type 2 Diabetes: A Patient-Centered Approach. Diabetes Care 2015, 38, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Colín-Lozano, B.; Estrada-Soto, S.; Chávez-Silva, F.; Gutiérrez-Hernández, A.; Cerón-Romero, L.; Giacoman-Martínez, A.; Almanza-Pérez, J.C.; Hernández-Núñez, E.; Wang, Z.; Xie, X.; et al. Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids. Molecules 2018, 23, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saidu, Y.; Muhammad, S.A.; Abbas, A.Y.; Onu, A.; Tsado, I.M.; Muhammad, L. In vitro screening for protein tyrosine phosphatase 1B and dipeptidyl peptidase IV inhibitors from selected Nigerian medicinal plants. J. Intercult. Ethnopharmacol. 2017, 6, 154–157. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: New insights and future directions. J. Med. Chem. 2015, 58, 2047–2067. [Google Scholar] [CrossRef]
- He, J.; Gao, H.-X.; Yang, N.; Zhu, X.-D.; Sun, R.-B.; Xie, Y.; Zheng, C.-H.; Zhang, J.-W.; Wang, J.-K.; Ding, F.; et al. The aldose reductase inhibitor epalrestat exerts nephritic protection on diabetic nephropathy in db/db mice through metabolic modulation. Acta Pharmacol. Sin. 2019, 40, 86–97. [Google Scholar] [CrossRef]
- Herrera-Rueda, M.A.; Tlahuext-Romero, H.; Paoli, P.; Giacoman-Martínez, A.; Almanza-Pérez, J.C.; Pérez-Sánchez, H.; Gutiérrez-Hernández, A.; Chávez-Silva, F.; Domínguez-Mendoza, E.A.; Estrada-Soto, S.; et al. Design, synthesis, in vitro, in vivo and in silico pharmacological characterization of antidiabetic N-Boc-L-tyrosine-based compounds. Biomed. Pharmacother. 2018, 108C, 670–678. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, S.; Navarrete-Vázquez, G.; Estrada-Soto, S.; Giles-Rivas, D.; Alarcón-Aguilar, F.J.; León-Rivera, I.; Giacoman-Martínez, A.; Miranda Pérez, E.; Almanza-Pérez, J.C. Discovery of New Dual PPARγ-GPR40 Agonists with Robust Antidiabetic Activity: Design, Synthesis and in Combo Drug Evaluation. Biomed. Pharmacother. 2017, 90, 53–61. [Google Scholar] [CrossRef]
- Maccari, R.; Del Corso, A.; Paoli, P.; Adornato, I.; Lori, G.; Balestri, F.; Cappiello, M.; Naß, A.; Wolber, G.; Ottanà, R. An investigation on 4-thiazolidinone derivatives as dual inhibitors of aldose reductase and protein tyrosine phosphatase 1B, in the search for potential agents for the treatment of type 2 diabetes mellitus and its complications. Bioorg Med. Chem. Lett. 2018, 28, 3712–3720. [Google Scholar] [CrossRef] [PubMed]
- Bialy, L.; Waldman, H. Inhibitors of Protein Tyrosine Phosphatases: Next-Generation Drug. Angew. Chem. Int. Edit. 2005, 44, 3814–3839. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, S.; Estrada-Soto, S.; Paoli, P.; Lori, G.; León-Rivera, I.; Navarrete-Vázquez, G. Synthesis and evaluation of thiazolidine-2,4-dione/benzazole derivatives as inhibitors of protein tyrosine phosphatase 1B (PTP-1B): Antihyperglycemic activity with molecular docking study. Biomed. Pharmacother. 2018, 107C, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA 2005, 102, 6679–6685. [Google Scholar] [CrossRef] [Green Version]
- Roy, U. 3D Modeling of Tumor Necrosis Factor Receptor and Tumor Necrosis Factor-bound Receptor Systems. Mol. Inform. 2019, 38, e1800011. [Google Scholar] [CrossRef] [PubMed]
- Roy, U. Structural modeling of tumor necrosis factor: A protein of immunological importance. Biotechnol. Appl. Biochem. 2017, 64, 454–463. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Y.-X.; Yi, K.-H.; Li, M.-Q.; Li, J.-Z.; Ye, F. Quantitative Structure Activity Relationship Studies and Molecular Dynamics Simulations of 2-(Aryloxyacetyl)cyclohexane-1,3-Diones Derivatives as 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors. Front. Chem. 2019, 7, 556. [Google Scholar] [CrossRef] [Green Version]
- Decherchi, S.; Cavalli, A. Thermodynamics and Kinetics of Drug-Target Binding by Molecular Simulation. Chem. Rev. 2020, 120, 12788–12833. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Costantino, L. Molecular dynamics simulations of the structure of aldose reductase complexed with the inhibitor tolrestat. Bioorg Med. Chem. Lett. 1998, 8, 641–646. [Google Scholar] [CrossRef]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. Correction to “admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties”. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Román-Ramos, R.; Alarcón-Aguilar, F.J.; Hernández-Rosado, J.V.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Navarrete-Vazquez, G. Discovery of thiazolidine-2,4-dione/biphenylcarbonitrile Hybrid as Dual PPAR α/γ Modulator With Antidiabetic Effect: In Vitro, in Silico and in Vivo Approaches. Chem. Biol. Drug. Des. 2013, 81, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Pharaon, L.F.; El-Orabi, N.F.; Kunhi, M.; Al-Yacoub, N.; Awad, S.M.; Poizat, C. Rosiglitazone promotes cardiac hypertrophy and alters chromatin remodeling in isolated cardiomyocytes. Toxicol. Lett. 2017, 280, 151–158. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, M.; Jafari, S.; Sajedi, B.; Gohari, S.; Akbarieh, S.; Heydari, A.H.; Jameshoorani, M. Comparison of fenofibrate and pioglitazone effects on patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2017, 29, 1385–1388. [Google Scholar] [CrossRef]
- Skochko, O.V.; Kaidashev, I.P. Effect of Pioglitazone on Insulin Resistance, Progression of Atherosclerosis and Clinical Course of Coronary Heart Disease. Wiad. Lek. 2017, 70, 881–890. [Google Scholar]
- American Diabetes Association, Diabetes Type 2. Available online: http://www.diabetes.org/diabetes-basics/type-2 (accessed on 15 September 2018).
- Numonov, S.; Edirs, S.; Bobakulov, K.; Nasimullah, M.Q.; Bozorov, K.; Sharopov, F.; Setzer, W.N.; Zhao, H.; Habasi, M.; Sharofova, M.; et al. Evaluation of the antidiabetic activity and chemical composition of Geranium collinum root extracts—Computational and experimental investigations. Molecules 2017, 22, 983. [Google Scholar] [CrossRef] [PubMed]
- Rotinsulu, H.; Yamazaki, H.; Sugai, S.; Iwakura, N.; Wewengkang, D.S.; Sumilat, D.A.; Namikoshi, M. Cladosporamide A, a New Protein Tyrosine Phosphatase 1B Inhibitor, Produced by an Indonesian Marine Sponge-Derived Cladosporium Sp. J. Nat. Med. 2018, 72, 779–783. [Google Scholar] [CrossRef]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.-B.; Sharpe, A.H.; et al. Regulation of Insulin-Like Growth Factor Type I (IGF-I) Receptor Kinase Activity by Protein Tyrosine Phosphatase 1B (PTP-1B) and Enhanced IGF-I-Mediated Suppression of Apoptosis and Motility in PTP-1B-Deficient Fibroblasts. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhajed, S.S.; Chaskar, S.; Kshirsagar, S.K.; Haldar, G.M.; Kar Mahapatra, D. Rational Design and Synthesis of Some PPAR-γ Agonists: Substituted benzylideneamino-benzylidene-thiazolidine-2,4-diones. Comput. Biol. Chem. 2017, 67, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Servicio Nacional de Sanidad, Inocuidad y Calidad Agroalimentaria, Gobierno de México, Norma Oficial Mexicana: NOM-062-ZOO-1999 Especificaciones técnicas para la producción, cuidado y uso de los animales de laboratorio. Available online: https://www.gob.mx/senasica/documentos/nom-062-zoo-1999 (accessed on 30 September 2018).
- Procuraduría Federal de Protección al Ambiente, Gobierno de México, Norma oficial Mexicana: NOM-033-SAG/ZOO-20, Métodos para dar muerte a los animales domésticos y silvestres. Available online: https://www.gob.mx/profepa/documentos/norma-oficial-mexicana-nom-033-sag-zoo-2014-metodos-para-dar-muerte-a-los-animales-domesticos-y-silvestres (accessed on 30 September 2018).
- Guzmán-Ávila, R.; Flores-Morales, V.; Paoli, P.; Camici, G.; Ramírez-Espinosa, J.J.; Cerón-Romero, L.; Navarrete-Vázquez, G.; Hidalgo-Figueroa, S.; Ríos, M.Y.; Villalobos-Molina, R.; et al. Ursolic acid derivatives as potential antidiabetic agents: In vitro, in vivo, and in silico studies. Drug Dev. Res. 2018, 79, 70–80. [Google Scholar] [CrossRef]
- Tahara, A.; Matsuyama-Yokono, A.; Shibasaki, M. Effects of Antidiabetic Drugs in High-Fat Diet and Streptozotocin-Nicotinamide-Induced Type 2 Diabetic Mice. Eur. J. Pharmacol. 2011, 655, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Hernández, A.; Galván-Ciprés, Y.; Domínguez-Mendoza, E.A.; Aguirre-Vidal, Y.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Navarrete-Vázquez, G. Design, Synthesis, Antihyperglycemic Studies, and Docking Simulations of Benzimidazole-Thiazolidinedione Hybrids. J. Chem. 2019, 1650145. [Google Scholar] [CrossRef]
- Krishnan, N.; Krishnan, K.; Connors, C.R.; Choy, M.S.; Page, R.; Peti, W.; Van Aelst, L.; Shea, S.D.; Tonks, N.K. PTP1B inhibition suggests a therapeutic strategy for Rett syndrome. J. Clin. Investig. 2015, 125, 3163–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, F.X.; Cousido-Siah, A.; Porté, S.; Domínguez, M.; Crespo, I.; Rechlin, C.; Mitschler, A.; de Lera, Á.R.; Martín, M.J.; de la Fuente, J.Á.; et al. Structural Determinants of the Selectivity of 3-Benzyluracil-1-acetic Acids toward Human Enzymes Aldose Reductase and AKR1B10. ChemMedChem 2015, 10, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Bajusz, D.; Rácz, A.; Héberger, K. Comparison of Data Fusion Methods as Consensus Scores for Ensemble Docking. Molecules 2019, 24, 2690. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Lara, E.; Martínez-Conde, C.; Rosales-Ortega, E.; Ramírez-Espinosa, J.J.; Rivera-Leyva, J.C.; Centurión, D.; Carvajal, K.; Ortega-Cuellar, D.; Estrada-Soto, S.; Navarrete-Vázquez, G. Synthesis and In Vitro AMPK Activation of Cycloalkyl/Alkarylbiguanides with Robust In Vivo Antihyperglycemic Action. J. Chem. 2017, 1212609. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.; Ahmad, R.; Azad, I.; Raza, S.; Joshi, S.; Khan, A.R. Computer-aided drug design and virtual screening of targeted combinatorial libraries of mixed-ligand transition metal complexes of 2-butanone thiosemicarbazone. Comput. Biol. Chem. 2018, 75, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Nava, E.J.; Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Arreguín-Espinosa, R.; Ortega-Cuellar, D.; González-Valdez, A.; Martínez-Rosas, V.; Morales-Luna, L.; Martínez-Miranda, J.; Sierra-Palacios, E.; et al. Novel inhibitors of human glucose-6-phosphate dehydrogenase (HsG6PD) affect the activity and stability of the protein. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129828. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Time Course and % of Variation of Glycemia | |||
|---|---|---|---|---|
| 1 h | 3 h | 5 h | 7 h | |
| 1 | 44.4 | 35.9 | 30.5 | 8.2 |
| 2 | −0.2 * | −22.8 * | −24.5 | −21.6 |
| 3 | 16.3 | −8.4 | −13.7 | −17.3 |
| 4 | 14.7 | 7.9 | −2.7 | −10.4 |
| 5 | 24.7 | −21.1 * | −14.1 | −16.1 |
| 6 | 7.1 * | −18.5 * | −18.5 | −30.0 |
| 7 | 20.6 | 19.7 | −1.6 | −8.8 |
| 8 | −3.1 * | −5.9 | −8.1 | −31.7 |
| 9 | 3.2 * | −10.7 * | −23.4 | −30.9 |
| Control | 52.9 | 15.3 | 0.2 | −11.5 |
| Glibenclamide | −12.8 * | −33.9 * | −42.6 * | −64.6 * |
| Property/Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Physicochemical properties | |||||||||
| Log P | |||||||||
| Predicted solubility | |||||||||
| Lipinski Ro5 | |||||||||
| Pharmacodynamics properties (Molecular docking) | |||||||||
| PTP-1B | |||||||||
| AR | |||||||||
| Pharmacokinetic properties | |||||||||
| Absorption | |||||||||
| PGp substrate | |||||||||
| CYP1A2 inhibitor | |||||||||
| CYP2D6 inhibitor | |||||||||
| CYP3A4 Inhibitor | |||||||||
| Toxicity prediction | |||||||||
| Mutagenicity | |||||||||
| Carcinogenicity | |||||||||
| hERG blockage | |||||||||
| Experimental antihyperglycemic data for analogue compounds [5,17] | |||||||||
| In vivo assay | No | No | No | No | Active | Active | No | Active | Active |
| Overall Score | |||||||||
| Predicted analysis result | |||||||||
| Current experimental result | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez-Mendoza, E.A.; Galván-Ciprés, Y.; Martínez-Miranda, J.; Miranda-González, C.; Colín-Lozano, B.; Hernández-Núñez, E.; Hernández-Bolio, G.I.; Palomino-Hernández, O.; Navarrete-Vazquez, G. Design, Synthesis, and In Silico Multitarget Pharmacological Simulations of Acid Bioisosteres with a Validated In Vivo Antihyperglycemic Effect. Molecules 2021, 26, 799. https://doi.org/10.3390/molecules26040799
Domínguez-Mendoza EA, Galván-Ciprés Y, Martínez-Miranda J, Miranda-González C, Colín-Lozano B, Hernández-Núñez E, Hernández-Bolio GI, Palomino-Hernández O, Navarrete-Vazquez G. Design, Synthesis, and In Silico Multitarget Pharmacological Simulations of Acid Bioisosteres with a Validated In Vivo Antihyperglycemic Effect. Molecules. 2021; 26(4):799. https://doi.org/10.3390/molecules26040799
Chicago/Turabian StyleDomínguez-Mendoza, Elix Alberto, Yelzyn Galván-Ciprés, Josué Martínez-Miranda, Cristian Miranda-González, Blanca Colín-Lozano, Emanuel Hernández-Núñez, Gloria I. Hernández-Bolio, Oscar Palomino-Hernández, and Gabriel Navarrete-Vazquez. 2021. "Design, Synthesis, and In Silico Multitarget Pharmacological Simulations of Acid Bioisosteres with a Validated In Vivo Antihyperglycemic Effect" Molecules 26, no. 4: 799. https://doi.org/10.3390/molecules26040799
APA StyleDomínguez-Mendoza, E. A., Galván-Ciprés, Y., Martínez-Miranda, J., Miranda-González, C., Colín-Lozano, B., Hernández-Núñez, E., Hernández-Bolio, G. I., Palomino-Hernández, O., & Navarrete-Vazquez, G. (2021). Design, Synthesis, and In Silico Multitarget Pharmacological Simulations of Acid Bioisosteres with a Validated In Vivo Antihyperglycemic Effect. Molecules, 26(4), 799. https://doi.org/10.3390/molecules26040799