
Chemical Profiles and Pharmacological Properties with in Silico Studies on Elatostema papillosum Wedd


 ,
,  ,
,  , ,
, ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
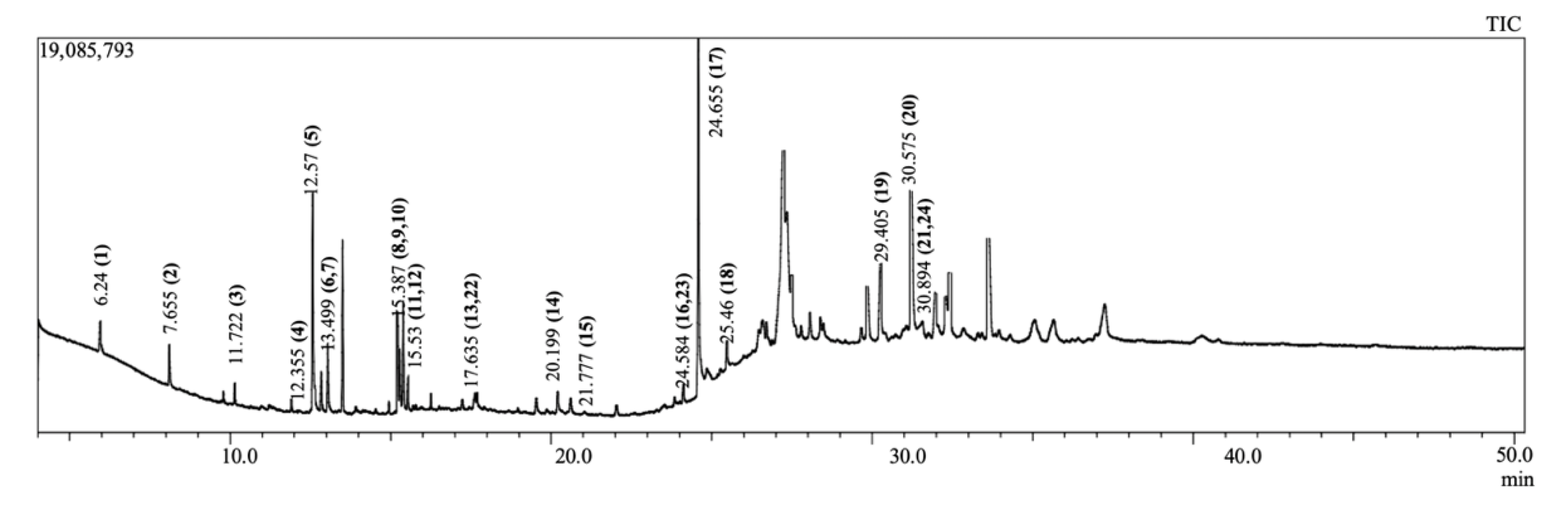
2.1. GC-MS Analysis
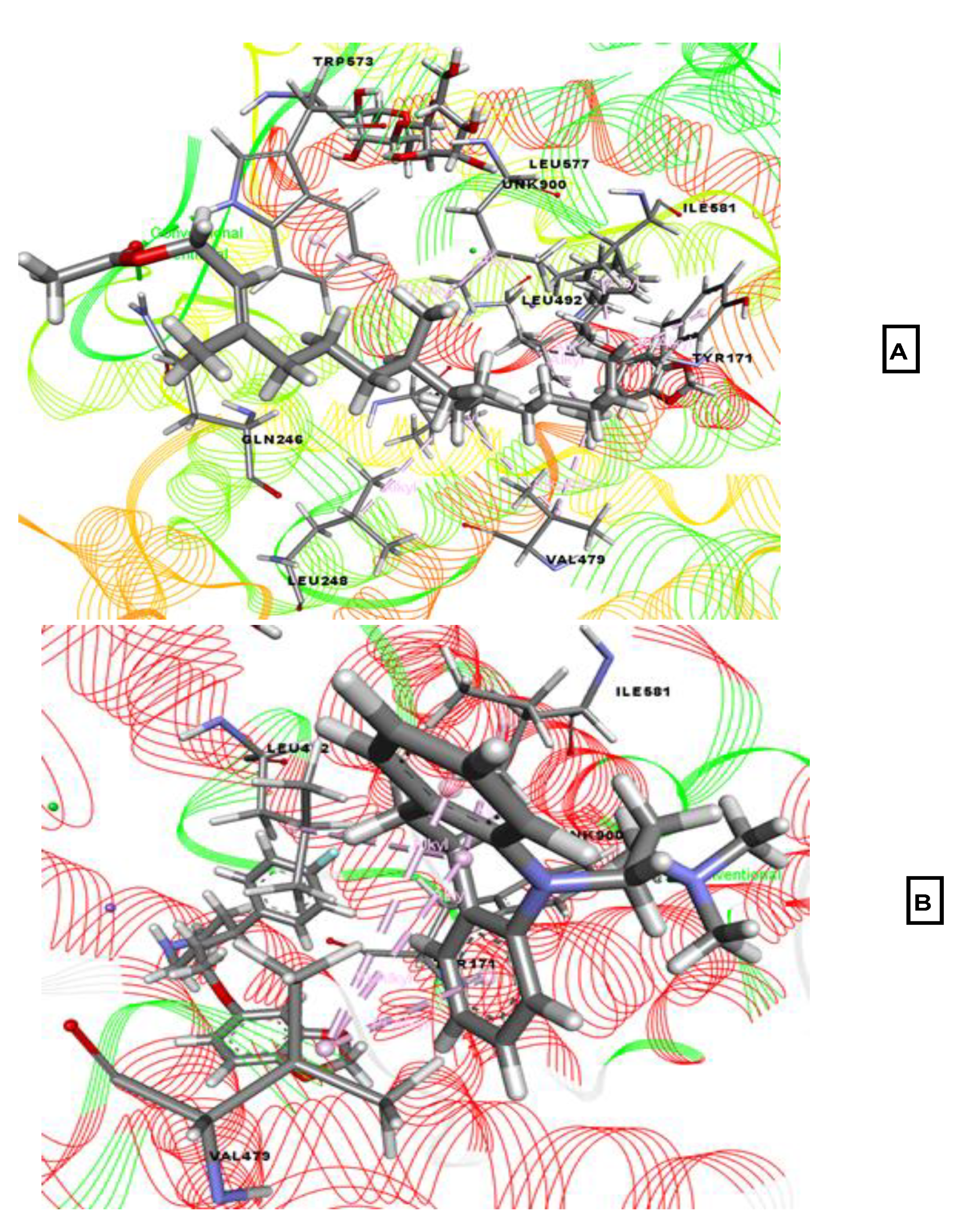
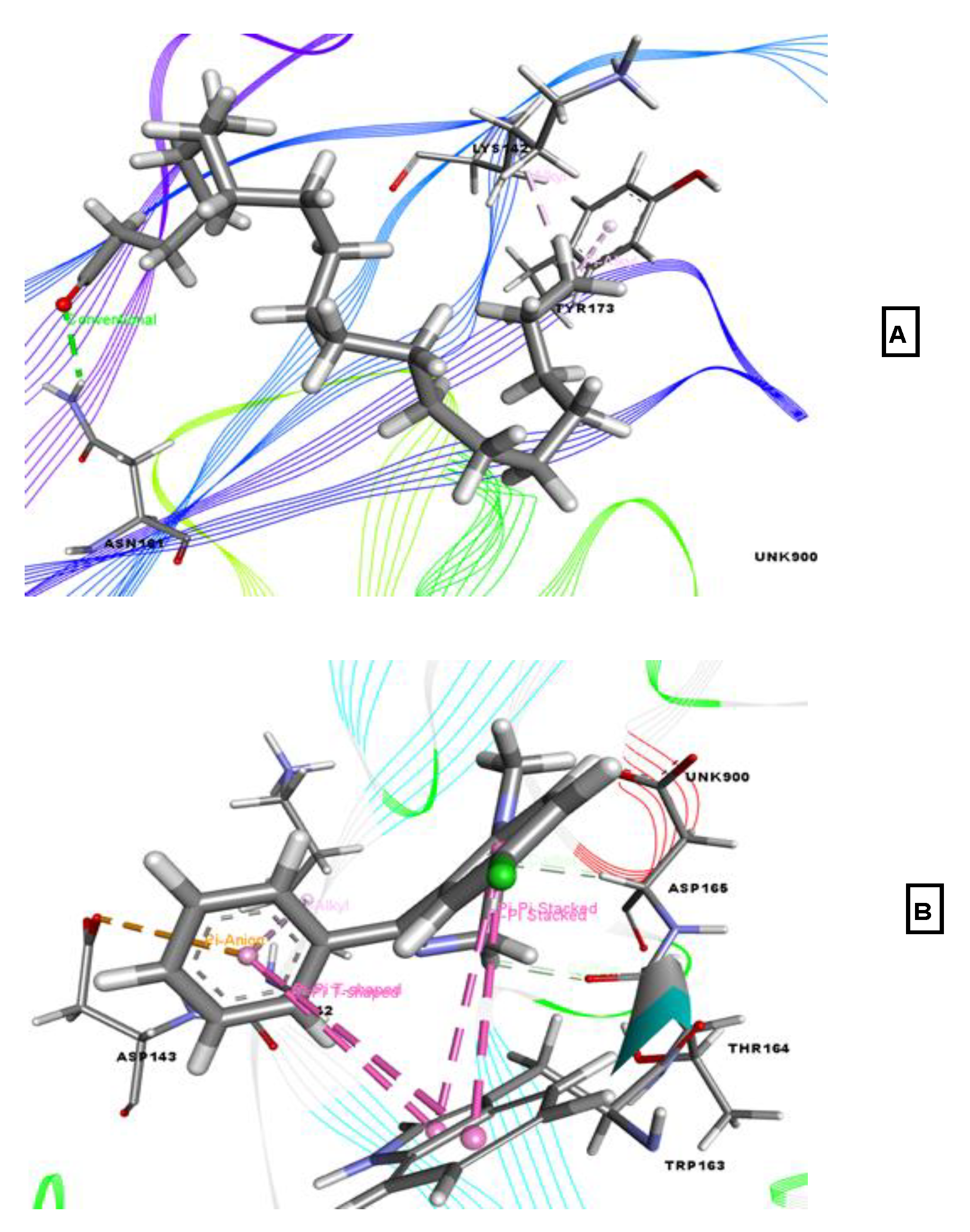
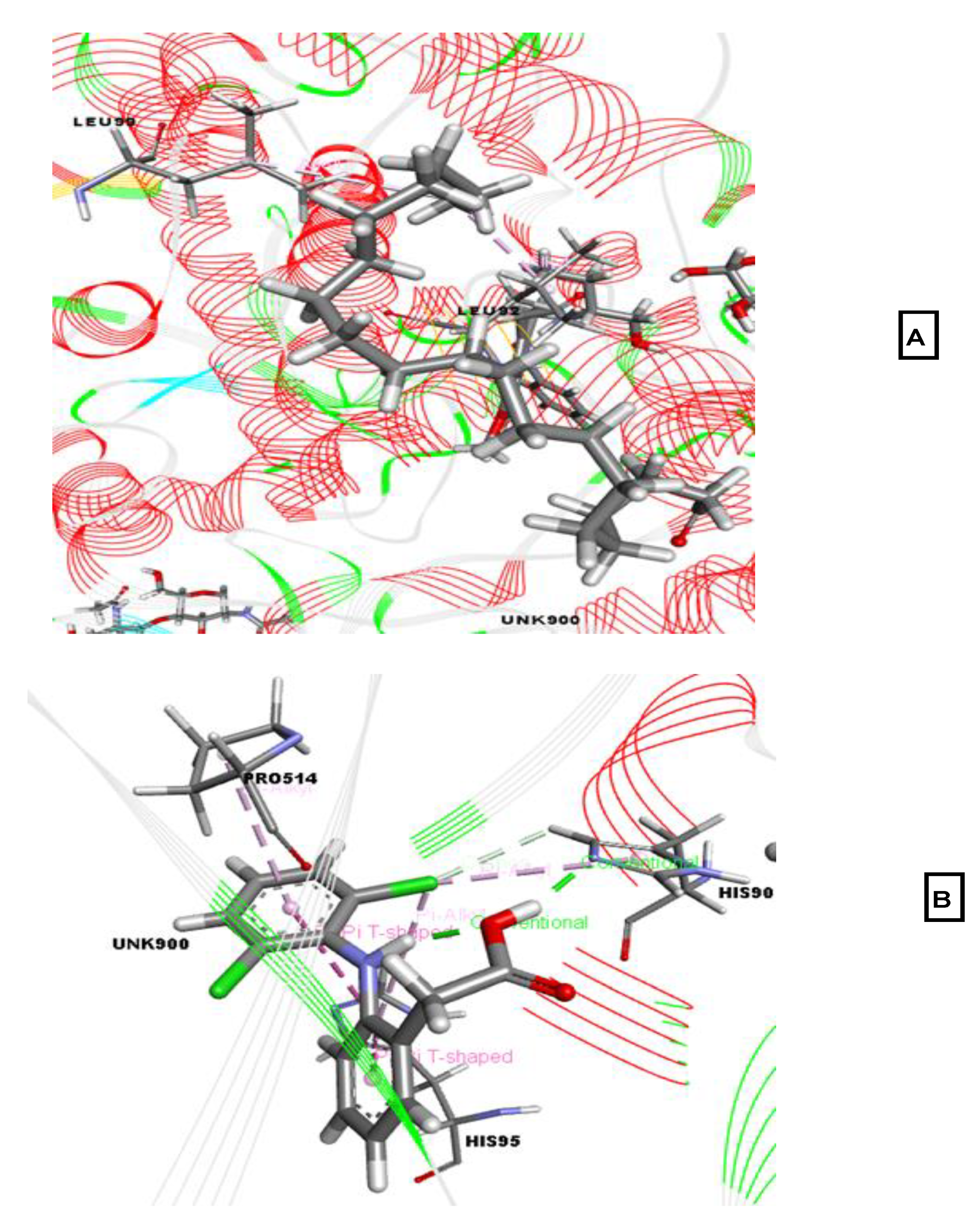
2.2. Molecular Docking Associated with Antidepressant Activity
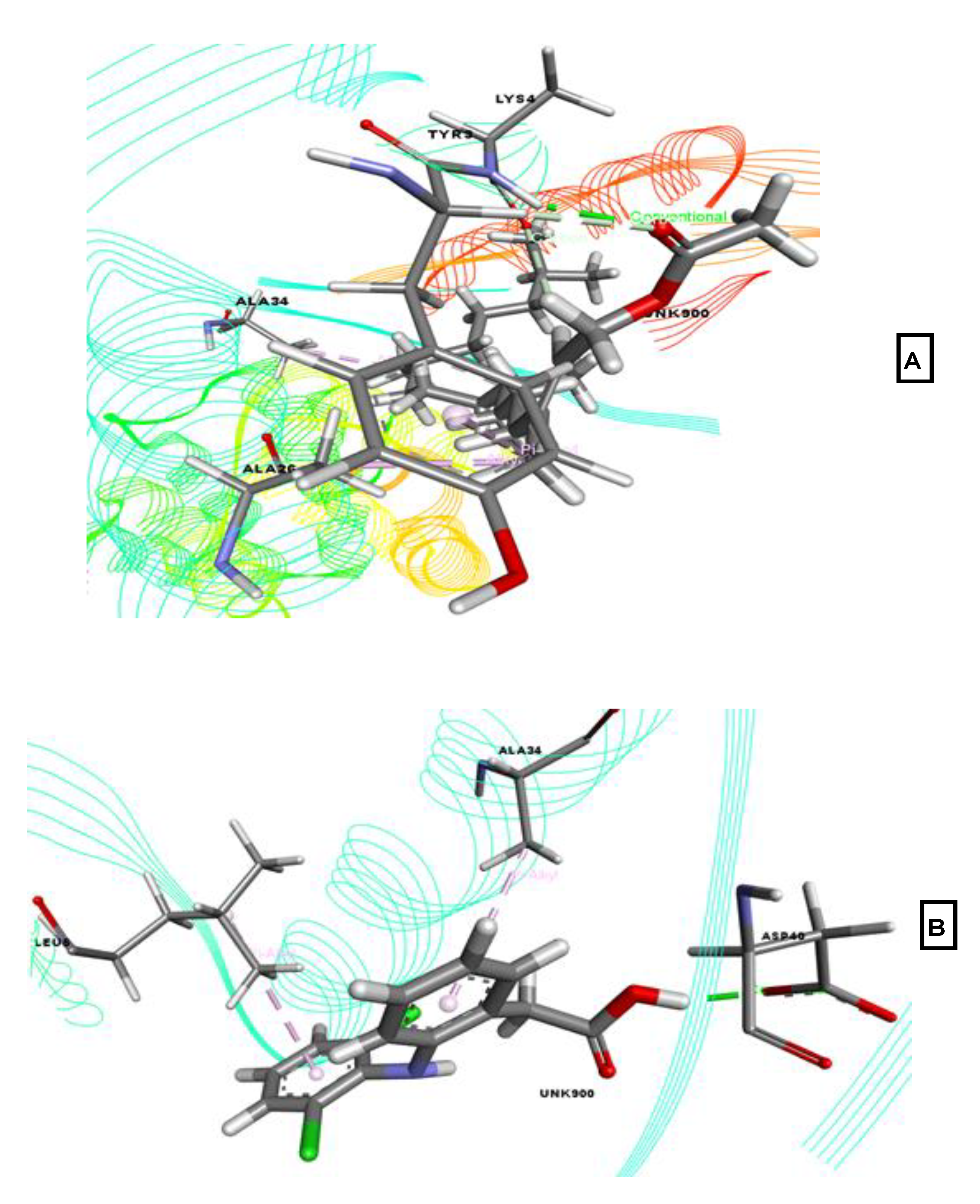
2.3. Molecular Docking Associated with Anxiolytic Activity
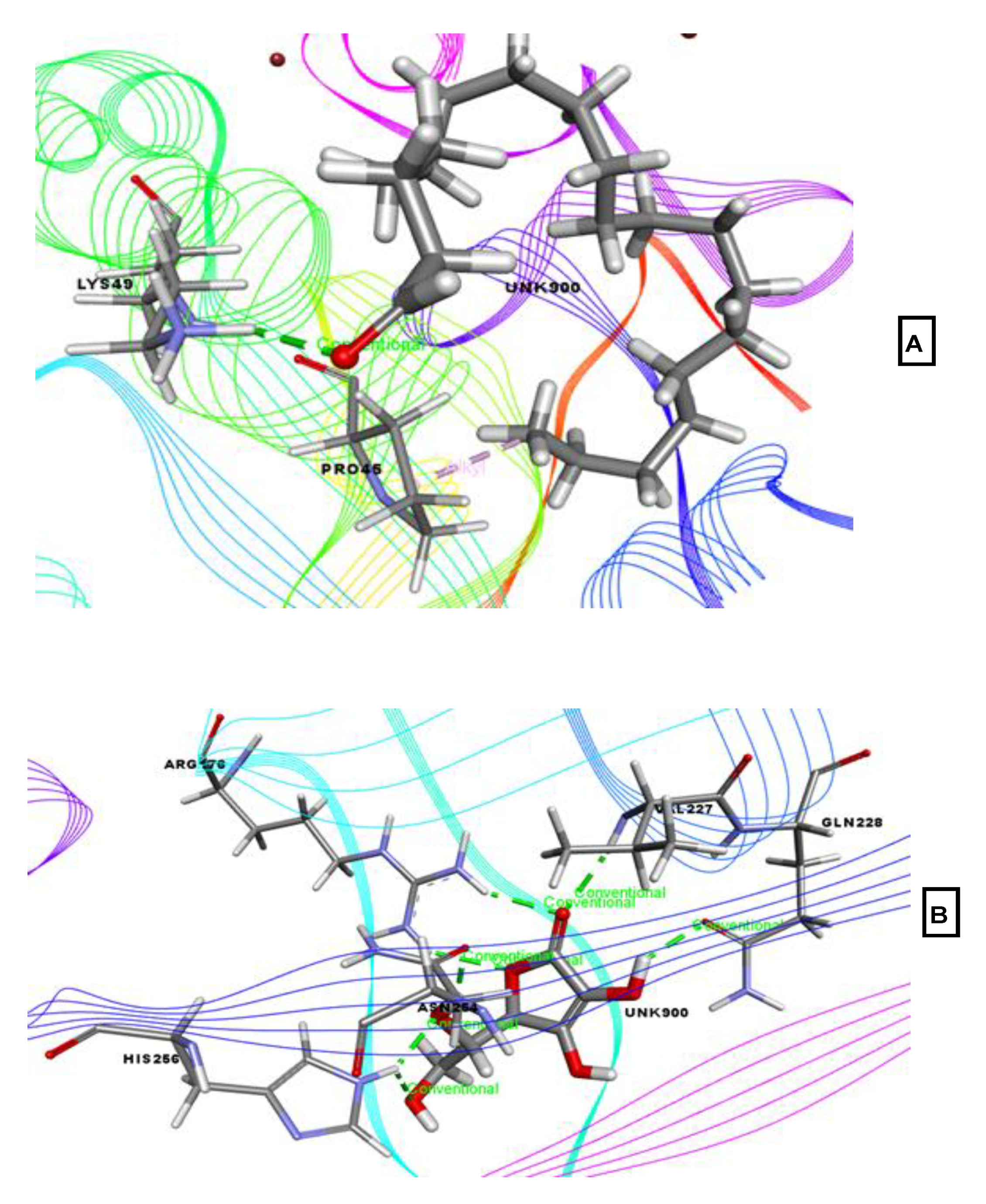
2.4. Molecular Docking Associated with Analgesic Activity
2.5. Molecular Docking Associated with Anti-Inflammatory Activity
2.6. Molecular Docking Associated with Antioxidant Activity
2.7. Ligand-Based ADME/T Predictions
2.8. Molecular Mechanics Generalized Born Surface Area (MM-GBSA) Analysis and Ligand Efficiency
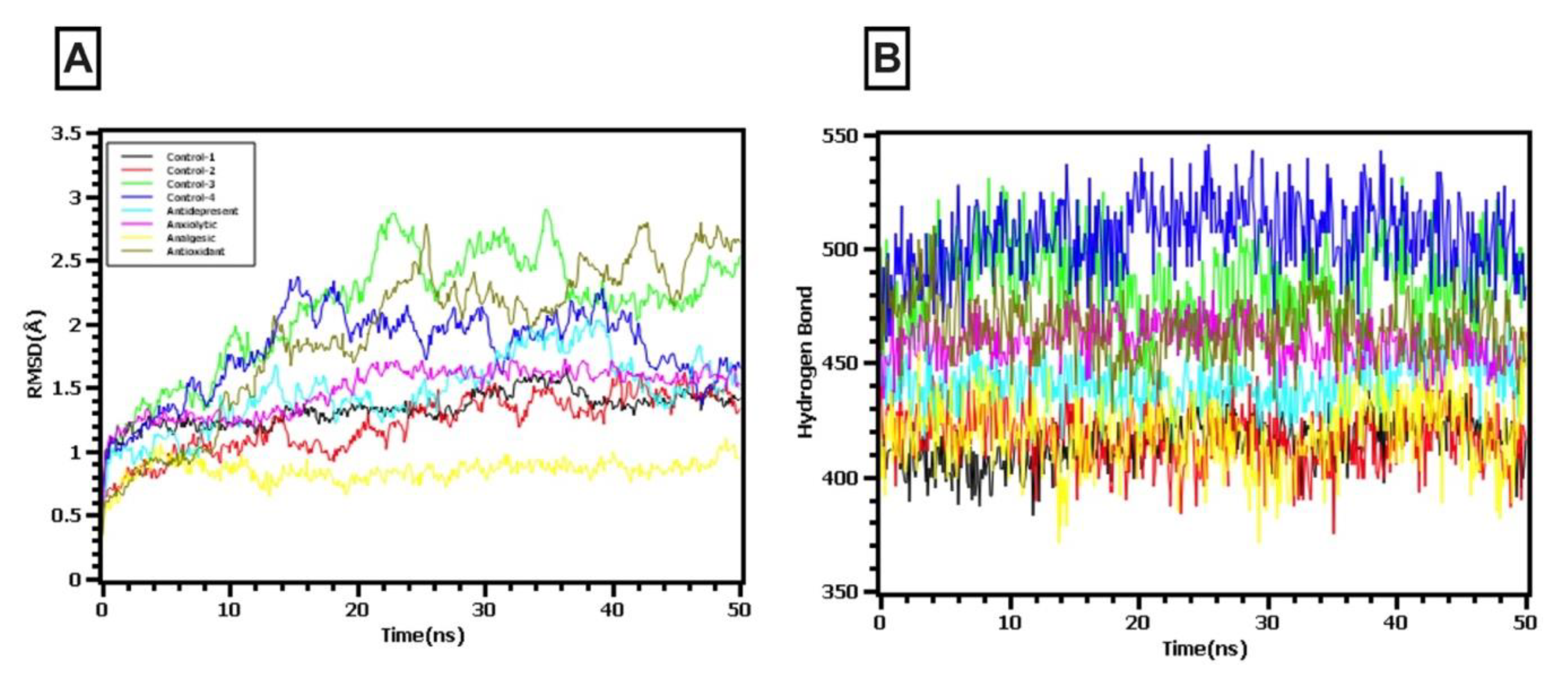
2.9. Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Preparation of Crude Methanolic Extract
4.2. Gas Chromatography-Mass Spectrometry (GC-MS) Analysis
4.3. Computational Molecular Docking Analysis
4.3.1. Chemical Compounds Studied
4.3.2. Preparation of Ligand
4.3.3. Preparation of Receptor/Enzymes
4.3.4. Glide Docking
4.4. Ligand-Based Pharmacokinetic Parameter Analysis
4.5. Prime Molecular Mechanics Generalized Born Surface Area (MM-GBSA) and Ligand Efficiency
4.6. Computational Molecular Dynamic Simulations Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mamun-Or-Rashid, M.; Islam, M.A.; Idris, T.; Amran, M.S. CNS-Antidepressant Activity of Crude Extracts and Different Fractions of Stem Bark of Acacia Nilotica. Pharm. Sci. Technol. 2017, 1, 13. [Google Scholar]
- Reynolds, E. Brain and mind: A challenge for WHO. Lancet 2003, 9373, 1924–1925. [Google Scholar] [CrossRef]
- Al Mahmud, Z.; Emran, T.B.; Qais, N.; Bachar, S.C.; Sarker, M.; Uddin, M.M.N. Evaluation of analgesic, anti-inflammatory, thrombolytic and hepatoprotective activities of roots of Premna esculenta (Roxb). J. Basic Clin. Physiol. Pharm. 2016, 27, 63–70. [Google Scholar] [CrossRef]
- Aziz, M.A.I.; Barua, N.; Tareq, A.M.; Alam, N.; Prova, R.J.; Mamun, M.N.; Sayeed, M.A.; Chowdhury, M.A.U.; Emran, T.B. Possible neuropharmacological effects of Adenia trilobata (Roxb.) in the Swiss Albino mice model. Future J. Pharm. Sci. 2020, 6, 1–8. [Google Scholar]
- Jyoti, M.A.; Barua, N.; Hossain, M.S.; Hoque, M.; Bristy, T.A.; Mahmud, S.; Kamruzzaman; Adnan, M.; Chy, M.N.U.; Paul, A.; et al. Unravelling the biological activities of the Byttneria pilosa leaves using experimental and computational approaches. Molecules 2020, 25, 4737. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.M.; Rakib, A.; Mitra, S.; Tareq, A.T.; Emran, T.B.; Ud-Daula, S.A.F.M.; Amin, M.N.; Simal-Gandara, J. The Genus Curcuma and Inflammation: Overview of the Pharmacological Perspectives. Plants 2021, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Banu, N.; Alam, N.; Islam, M.N.; Islam, S.; Sakib, S.A.; Hanif, N.B.; Chowdhury, M.R.; Tareq, A.M.; Chowdhury, K.H.; Jahan, S.; et al. Insightful Valorization on Biological Activities of Pani Heloch Leaves through Experimental and Computer-Aided Mechanisms. Molecules 2020, 25, 5153. [Google Scholar] [CrossRef] [PubMed]
- Saxena, G.; McCutcheon, A.; Farmer, S.; Towers, G.; Hancock, R. Antimicrobial constituents of Rhus glabra. J. Ethnopharmacol. 1994, 42, 95–99. [Google Scholar] [CrossRef]
- Calixto, J.B.; Scheidt, C.; Otuki, M.; Santos, A.R. Biological activity of plant extracts: Novel analgesic drugs. Expert Opin. Emerg. Drugs 2001, 6, 261–279. [Google Scholar] [CrossRef]
- Akter, S.; Shah, M.; Tareq, A.M.; Nasrin, M.S.; Rahman, M.A.; Babar, Z.; Haque, M.A.; Royhan, M.J.; Mamun, M.N.; Reza, A.A. Pharmacological effect of methanolic and hydro-alcoholic extract of Coconut endocarp. J. Adv. Biotechnol. Exp. 2020, 3, 171–181. [Google Scholar] [CrossRef]
- Uddin, S.N. Traditional Uses of Ethnomedicinal Plants of the Chittagong Hill Tracts; Bangladesh National Herbarium: Dhaka, Bangladesh, 2006. [Google Scholar]
- Ali Reza, A.; Hossain, M.S.; Akhter, S.; Rahman, M.R.; Nasrin, M.S.; Uddin, M.J.; Sadik, G.; Khurshid Alam, A. In vitro antioxidant and cholinesterase inhibitory activities of Elatostema papillosum leaves and correlation with their phytochemical profiles: A study relevant to the treatment of Alzheimer’s disease. BMC Complement. Altern. Med. 2018, 18, 123. [Google Scholar] [CrossRef] [Green Version]
- Williamson, E.M.; Okpako, D.T.; Evans, F.J. Selection, Preparation and Pharmacological Evaluation of Plant Material; John Wiley & Sons: Hoboken, NJ, USA, 1996; Volume 1. [Google Scholar]
- Tareq, A.M.; Sohel, M.; Uddin, M.; Mahmud, M.H.; Hoque, M.; Reza, A.A.; Nasrin, M.S.; Kader, F.B.; Emran, T.B. Possible neuropharmacological effects of Apis cerana indica beehive in the Swiss Albino mice. JABET 2020, 3, 128–134. [Google Scholar]
- Barua, N.; Aziz, M.A.I.; Tareq, A.M.; Sayeed, M.A.; Alam, N.; ul Alam, N.; Uddin, M.A.; Lyzu, C.; Emran, T.B. In vivo and in vitro evaluation of pharmacological activities of Adenia trilobata (Roxb.). Biochem. Biophys. Rep. 2020, 23, 100772. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.-E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Rakib, A.; Ahmed, S.; Islam, M.A.; Uddin, M.M.N.; Paul, A.; Chy, M.N.U.; Emran, T.B.; Seidel, V. Pharmacological studies on the antinociceptive, anxiolytic and antidepressant activity of Tinospora crispa. Phytother. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Kumar, B.; Kaur, M.; Kaur, G.; Kaur, H. Phytochemical screening and extraction: A review. Int. Pharm. Sci. 2011, 1, 98–106. [Google Scholar]
- Uddin, M.Z.; Emran, T.B.; Dutta, M.; Ullah, S.A.; Rana, S.H.M.S. In vivo antidepressant, analgesic, anti-inflammatory activities, in vitro antioxidant and antibacterial potential of fractionated Elatostema papillosum Wed. extract. Pharma Innov. J. 2019, 8, 241–246. [Google Scholar]
- Rahman, M.A.; bin Imran, T.; Islam, S. Antioxidative, antimicrobial and cytotoxic effects of the phenolics of Leea indica leaf extract. Saudi J. Biol. Sci. 2013, 20, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Rakib, A.; Ahmed, S.; Islam, M.A.; Haye, A.; Uddin, S.N.; Uddin, M.M.N.; Hossain, M.K.; Paul, A.; Emran, T.B. Antipyretic and hepatoprotective potential of Tinospora crispa and investigation of possible lead compounds through in silico approaches. Food Sci. Nutr. 2020, 8, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Rahman, J.; Tareq, A.M.; Hossain, M.; Sakib, S.A.; Islam, M.N.; Ali, M.; Uddin, A.; Hoque, M.; Nasrin, M.; Emran, T.B. Biological Evaluation, DFT Calculations and Molecular Docking Studies on the Antidepressant and Cytotoxicity Activities of Cycas pectinata Buch.-Ham. Compounds. Pharmaceuticals 2020, 13, 232. [Google Scholar] [CrossRef]
- Bristy, T.A.; Barua, N.; Montakim Tareq, A.; Sakib, S.A.; Etu, S.T.; Chowdhury, K.H.; Jyoti, M.A.; Aziz, M.; Ibn, A.; Reza, A. Deciphering the pharmacological properties of methanol extract of Psychotria calocarpa leaves by in vivo, in vitro and in silico approaches. Pharmaceuticals 2020, 13, 183. [Google Scholar] [CrossRef]
- Dutta, T.; Paul, A.; Majumder, M.; Sultan, R.A.; Emran, T.B. Pharmacological evidence for the use of Cissus assamica as a medicinal plant in the management of pain and pyrexia. Biochem. Biophys. Rep. 2020, 21, 100715. [Google Scholar] [CrossRef]
- Jahan, I.; Tona, M.R.; Sharmin, S.; Sayeed, M.A.; Tania, F.Z.; Paul, A.; Chy, M.; Uddin, N.; Rakib, A.; Emran, T.B. GC-MS phytochemical profiling, pharmacological properties, and in silico studies of Chukrasia velutina leaves: A novel source for bioactive agents. Molecules 2020, 25, 3536. [Google Scholar] [CrossRef]
- Khan, S.; Nazir, M.; Raiz, N.; Saleem, M.; Zengin, G.; Fazal, G.; Saleem, H.; Mukhtar, M.; Tousif, M.I.; Tareen, R.B. Phytochemical profiling, in vitro biological properties and in silico studies on Caragana ambigua stocks (Fabaceae): A comprehensive approach. Ind. Crops Prod. 2019, 131, 117–124. [Google Scholar] [CrossRef]
- Adnan, M.; Chy, N.U.; Mostafa Kamal, A.; Azad, M.O.K.; Paul, A.; Uddin, S.B.; Barlow, J.W.; Faruque, M.O.; Park, C.H.; Cho, D.H. Investigation of the biological activities and characterization of bioactive constituents of Ophiorrhiza rugosa var. prostrata (D. Don) & Mondal leaves through in vivo, in vitro, and in silico approaches. Molecules 2019, 24, 1367. [Google Scholar]
- Ahmed, S.; Rakib, A.; Islam, M.A.; Khanam, B.H.; Faiz, F.B.; Paul, A.; Chy, M.N.U.; Bhuiya, N.M.A.; Uddin, M.M.N.; Ullah, S.A. In vivo and in vitro pharmacological activities of Tacca integrifolia rhizome and investigation of possible lead compounds against breast cancer through in silico approaches. Clin. Phytosci. 2019, 5, 36. [Google Scholar] [CrossRef]
- Grosser, T.; Theken, K.N.; FitzGerald, G.A. Cyclooxygenase inhibition: Pain, inflammation, and the cardiovascular system. Clin. Pharm. 2017, 102, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Hasanat, A.; Chowdhury, T.A.; Kabir, M.S.H.; Chowdhury, M.S.; Chy, M.; Uddin, N.; Barua, J.; Chakrabarty, N.; Paul, A. Antinociceptive Activity of Macaranga denticulata Muell. Arg. (Family: Euphorbiaceae): In Vivo and In Silico Studies. Medicines 2017, 4, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tareq, A.M.; Farhad, S.; Uddin, A.N.; Hoque, M.; Nasrin, M.S.; Uddin, M.M.R.; Hasan, M.; Sultana, A.; Munira, M.S.; Lyzu, C. Chemical profiles, pharmacological properties, and in silico studies provide new insights on Cycas pectinata. Heliyon 2020, 6, e04061. [Google Scholar] [CrossRef] [PubMed]
- Krisnamurti, G.; Fatchiyah, F. Interaction of acetaminophen and caffeine towards cyclooxygenase-2 (COX-2) in inhibition of prostaglandin (PGH 2) synthesis. JPhCS 2019, 1146, 012004. [Google Scholar]
- Baeuerle, P.A.; Baichwal, V.R. NF-kB as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv. Immunol. 1997, 65, 111–138. [Google Scholar] [PubMed]
- Xu, L.; Liu, Y.; Sun, Y.; Li, H.; Mi, W.; Jiang, Y. Analgesic effects of TLR4/NF-κB signaling pathway inhibition on chronic neuropathic pain in rats following chronic constriction injury of the sciatic nerve. Biomed. Pharm. 2018, 107, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.S.S.; Silva, H.B.F.; Rios, R.; de Oliveira, A.P.; Carneiro, N.V.Q.; Costa, R.S.; Alves, W.S.; Souza, F.-L.M.; da Silva Velozo, E.; de Souza, S.A. The anti-allergic activity of Cymbopogon citratus is mediated via inhibition of nuclear factor kappa B (NF-ΚB) activation. BMC Complement. Altern. Med. 2015, 15, 168. [Google Scholar]
- Joseph, A.; Thuy, T.T.T.; Thanh, L.T.; Okada, M. Antidepressive and anxiolytic effects of ostruthin, a TREK-1 channel activator. PLoS ONE 2018, 13, e0201092. [Google Scholar] [CrossRef] [Green Version]
- Kubacka, M.; Mogilski, S.; Bednarski, M.; Nowiński, L.; Dudek, M.; Żmudzka, E.; Siwek, A.; Waszkielewicz, A.M.; Marona, H.; Satała, G. Antidepressant-like activity of aroxyalkyl derivatives of 2-methoxyphenylpiperazine and evidence for the involvement of serotonin receptor subtypes in their mechanism of action. Pharm. Biochem. Behav. 2016, 141, 28–41. [Google Scholar] [CrossRef]
- Adnan, M.; Chy, M.; Uddin, N.; Kama, A.; Azad, M.; Kalam, O.; Chowdhury, K.A.A.; Kabir, M.S.H.; Gupta, S.D.; Chowdhury, M. Comparative Study of Piper sylvaticum Roxb. Leaves and Stems for Anxiolytic and Antioxidant Properties Through in vivo, in vitro, and in silico Approaches. Biomedicines 2020, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Bharathi, R.; Santhi, N. 4-(3-(2-amino-3, 5-dibromophenyl)-1-(4-substitutedbenzoyl)-4, 5-dihydro-1H-pyrazol-5-yl) benzonitrile as a novel anti-Inflammatory scaffold: Synthesis, biological evaluation and docking studies. World Sci. News 2019, 126, 148–162. [Google Scholar]
- Duffy, F.J.; Devocelle, M.; Shields, D.C. Computational approaches to developing short cyclic peptide modulators of protein–protein interactions. In Computational Peptidology; Springer: New York, NY, USA, 2015; pp. 241–271. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Berman, H.; Battistuz, T.; Bhat, T.; Bluhm, W.; Bourne, P.; Burkhardt, K.; Feng, Z.; Gilliland, G.; Iype, L.; Jain, S. The protein data bank. Acta Crystallogr. D Biol. Cryst. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Lenaeus, M.J.; Burdette, D.; Wagner, T.; Focia, P.J.; Gross, A. Structures of KcsA in complex with symmetrical quaternary ammonium compounds reveal a hydrophobic binding site. Biochemistry 2014, 53, 5365–5373. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Yin, Q.; Wu, H. Structural studies of NF-κB signaling. Cell Res 2011, 21, 183–195. [Google Scholar] [CrossRef]
- Harman, C.A.; Turman, M.V.; Kozak, K.R.; Marnett, L.J.; Smith, W.L.; Garavito, R.M. 2007. Structural basis of enantioselective inhibition of cyclooxygenase-1 by S-alpha-substituted indomethacin ethanolamides. J. Biol. Chem. 2007, 282, 28096–28105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Stallings, W.C. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Chy, M.N.U.; Adnan, M.; Rauniyar, A.K.; Amin, M.M.; Majumder, M.; Islam, M.S.; Afrin, S.; Farhana, K.; Nesa, F.; Sany, M.A. Evaluation of anti-nociceptive and anti-inflammatory activities of Piper sylvaticum (Roxb.) stem by experimental and computational approaches. Orient. Pharm. Exp. Med. 2019, 1–15. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]
- Krieger, E. Yet another Scientific Artificial Reality Application (YASARA). 2004. Available online: http://www.yasara.org/ (accessed on 4 February 2021).
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical p K a prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Bappy, S.S.; Sultana, S.; Adhikari, J.; Mahmud, S.; Khan, M.A.; Kibria, K.M.K.; Rahman, M.M.; Shibly, A.Z. Extensive immunoinformatics study for the prediction of novel peptide-based epitope vaccine with docking confirmation against envelope protein of Chikungunya virus: A computational biology approach. J. Biomol. Struct. Dyn. 2020, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Swargiary, A.; Mahmud, S.; Saleh, M.A. Screening of phytochemicals as potent inhibitor of 3-chymotrypsin and papain-like proteases of SARS-CoV2: An in silico approach to combat COVID-19. J. Biomol. Struct. Dyn. 2020, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Mahmud, S.; Alam, A.S.M.R.U.; Rahman, M.E.; Ahmed, F.; Rahmatullah, M. Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: A molecular docking study. J. Biomol. Struct. Dyn. 2020, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]










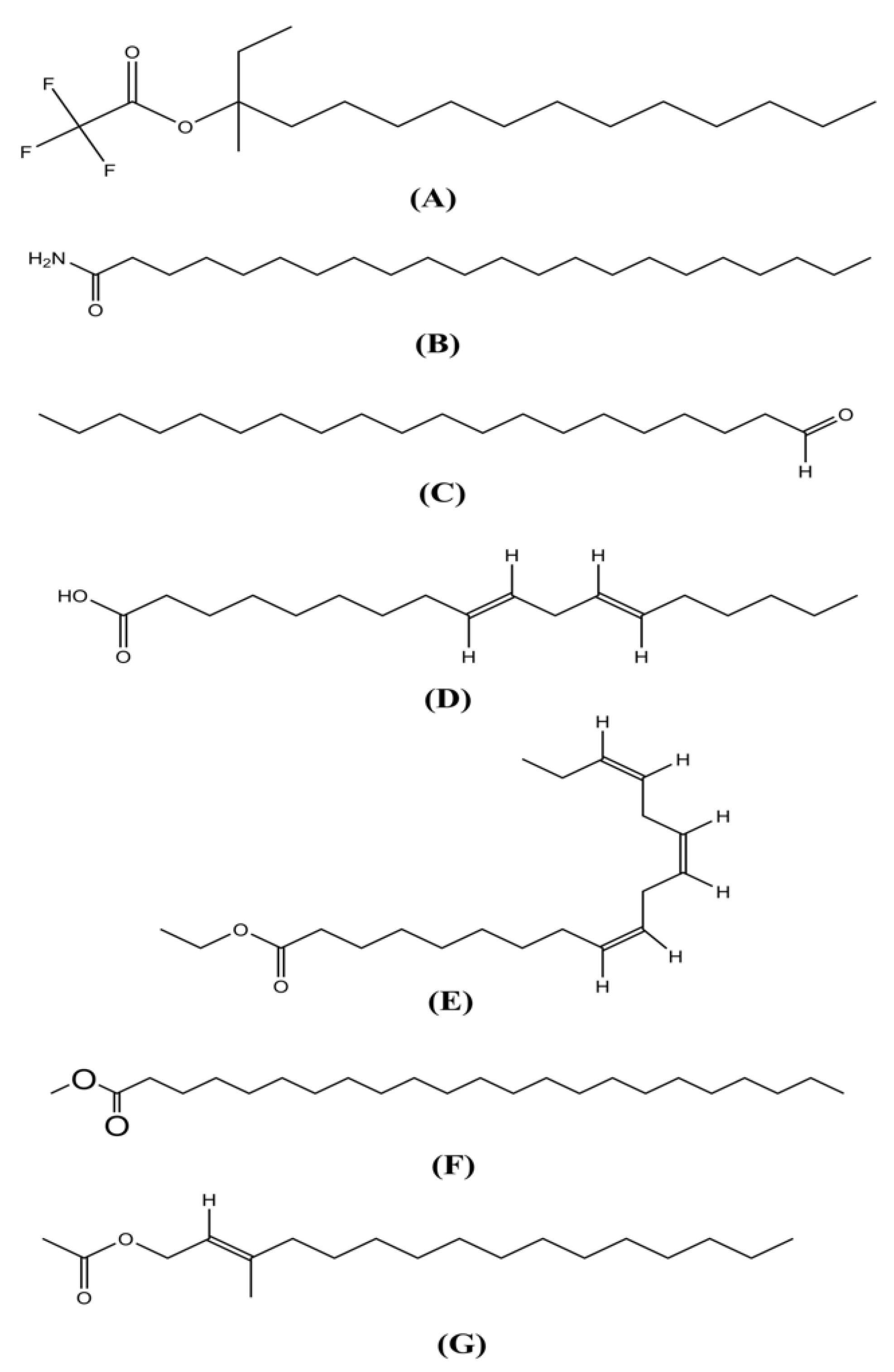{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl No. | Name | RT | m/z | Area | PA (%) | Molecular Formula | Nature |
|---|---|---|---|---|---|---|---|
| 1 | 3-butynoic acid | 6.24 | 40 | 53,887 | 0.282064878 | C4H4O2 | Monocarboxylic acid |
| 2 | Allene | 7.655 | 40 | 116,762 | 0.611176337 | C3H4 | Dienes |
| 3 | Sebacic acid | 11.722 | 40 | 36,746 | 0.19234242 | C16H34O4Si2 | Fatty acids |
| 4 | 9-octadecen-1-ol | 12.355 | 40 | 73,511 | 0.384784294 | C18H36O | Fatty alcohol |
| 5 | Phytol acetate | 12.57 | 81 | 705,294 | 3.691774752 | C22H42O2 | Diterpene |
| 6 | Hexadecanoic acid, methyl ester | 13.499 | 74 | 3,180,878 | 16.64991492 | C17H34O2 | Fatty acid |
| 7 | Pentadecanoic acid, 14-methyl-, methyl ester | 13.499 | 74 | 3,180,878 | 16.64991492 | C17H34O2 | Fatty acid |
| 8 | Linoleic acid ethyl ester | 15.387 | 44 | 90,498 | 0.473700657 | C₂₀H₃6O₂ | Fatty acid |
| 9 | Linoelaidic acid | 15.387 | 44 | 90,498 | 0.473700657 | C₁₈H₃₂O₂ | Fatty acid |
| 10 | Phytol | 15.387 | 44 | 90,498 | 0.473700657 | C20H40O | Diterpene |
| 11 | Heptacosanoic acid, methyl ester | 15.53 | 74 | 546,048 | 2.858221139 | C28H56O2 | Fatty acid |
| 12 | Tricosanoic acid methyl ester | 15.53 | 74 | 546,048 | 2.858221139 | C24H48O2 | Fatty acid |
| 13 | 3-trifluoroacetoxypentadecane | 17.635 | 44 | 63,831 | 0.334115524 | C17H31F3O2 | Ester |
| 14 | Hexadecanoic acid, 2-hydroxy-1-(hydroxymethyl)ethyl ester | 20.199 | 44 | 81,277 | 0.425434466 | C19H38O4 | Fatty acid glycerol ester |
| 15 | Epinephrine | 21.777 | 44 | 40,058 | 0.209678677 | C₉H₁₃NO₃ | Alkaloid |
| 16 | Cis-11-eicosenamide | 24.584 | 59 | 4,682,201 | 24.50840562 | C20H39NO | Amide |
| 17 | 2-(16-acetoxy-11-hydroxy-4,8,10,14-tetramethyl-3-oxohexadecahydrocyclopenta[a]phenanthren-17-ylidene)-6-methyl-hept-5-enoic acid, methyl ester | 24.655 | 207 | 160,560 | 0.840431585 | C32H48O6 | Fatty acid |
| 18 | 3-ethoxy-1,1,1,5,5,5-hexamethyl-3-(trimethylsiloxy)trisiloxane | 25.46 | 207 | 231,720 | 1.212909858 | C17H50O7Si7 | Silicate |
| 19 | 1,2-bis(trimethylsilyl)benzene | 29.405 | 207 | 37,161 | 0.194514687 | C12H22Si2 | Organic compound |
| 20 | Stigmasterol | 30.575 | 207 | 93,362 | 0.488691913 | C₂₉H₄₈O | Phytosterols |
| 21 | Beta-sitosterol | 30.894 | 207 | 126,310 | 0.66115417 | C29H50O | Phytosterols |
| 22 | Eicosanal | 17.635 | 44 | 67,933 | 0.355586939 | C20H40O | Aldehyde |
| 23 | 13-docosenamide | 24.584 | 59 | 4,682,201 | 24.50840562 | C22H43NO | Amide |
| 24 | Gamma-sitosterol | 30.894 | 207 | 126,310 | 0.66115417 | C29H50 O | Phytosteroids |
| Compounds Name | Docking Score (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| 5I6X | 4UUJ | COX-1 | COX-2 | 5LDE | 1HD2 | |
| 3-trifluoroacetoxypentadecane | −3.423 | −2.512 | −3.458 | - | −4.012 | −1.469 |
| 13-docosenamide | - | - | - | - | - | - |
| Linoelaidic acid | −0.797 | −0.265 | −0.410 | −2.960 | −1.194 | 1.170 |
| Linoleic acid ethyl ester | - | - | - | - | - | - |
| Eicosanal | −2.525 | −3.199 | −3.561 | - | 2.696 | −3.928 |
| Phytol acetate | −3.628 | −2.913 | −3.533 | −5.236 | −4.153 | −1.469 |
| Tricosanoic acid methyl ester | - | - | - | - | - | - |
| Standard (Imipramine HCl/ Diazepam/ Diclofenac-Na/ Ascorbic acid) | −5.350 | −4.035 | −4.590 | −7.260 | −5.758 | −5.134 |
| Compound Name | MW a | HB Donors b | HB Acceptors c | SASAd | QPlogPo/w e | QPlogBB f | QPlogS g | %Human Oral Absorption h |
|---|---|---|---|---|---|---|---|---|
| 3-trifluoroacetoxy pentadecane | 324.426 | 0 | 2 | 721.905 | 6.548 | −0.338 | −7.253 | 100 |
| 13-docosenamide | 337.588 | 2 | 3 | 864.813 | 5.197 | −0.461 | −9.339 | 88.609 |
| Linoelaidic acid | 280.45 | 1 | 2 | 717.95 | 5.83 | −1.523 | −6.373 | 90.462 |
| Linoleic acid ethyl ester | 308.503 | 0 | 3 | 670.733 | 4.826 | 0.25 | −7.357 | 100 |
| Eicosanal | 296.535 | 0 | 2 | 838.966 | 6.518 | −0.086 | −11.068 | 100 |
| Phytol acetate | 338.573 | 0 | 2 | 681.355 | 6.661 | −0.623 | −5.802 | 100 |
| Tricosanoic acid methyl ester | 368.642 | 1 | 3 | 977.143 | 7.528 | 0.138 | −13.069 | 100 |
| Compound Name/Standard | MM-GBSA ΔG Bind | Ligand Efficiency |
|---|---|---|
| Antidepressant Activity | ||
| Phytol acetate | −72.00 | 3.00 |
| Imipramine HCl (Standard) | −49.18 | 2.24 |
| Anxiolytic Activity | ||
| Eicosanal | −42.06 | 2.00 |
| Phenobarbital (Standard) | −30.65 | 1.80 |
| Anti-inflammatory Activity Cyclooxygenase-1 (COX-1) | ||
| Eicosanal | −47.86 | 2.28 |
| Diclofenac-Na (Standard) | −37.32 | 1.87 |
| Cyclooxygenase-2 (COX-2) | ||
| Phytol acetate | −75.25 | 3.14 |
| Diclofenac-Na (Standard) | −54.64 | 2.73 |
| NF-κB | ||
| Phytol acetate | −88.59 | 3.69 |
| Diclofenac-Na (Standard) | −52.91 | 2.65 |
| Antioxidant Activity | ||
| Eicosanal | −48.99 | 2.33 |
| Ascorbic acid (Standard) | −35.54 | 2.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uddin, M.Z.; Paul, A.; Rakib, A.; Sami, S.A.; Mahmud, S.; Rana, M.S.; Hossain, S.; Tareq, A.M.; Dutta, M.; Emran, T.B.; et al. Chemical Profiles and Pharmacological Properties with in Silico Studies on Elatostema papillosum Wedd. Molecules 2021, 26, 809. https://doi.org/10.3390/molecules26040809
Uddin MZ, Paul A, Rakib A, Sami SA, Mahmud S, Rana MS, Hossain S, Tareq AM, Dutta M, Emran TB, et al. Chemical Profiles and Pharmacological Properties with in Silico Studies on Elatostema papillosum Wedd. Molecules. 2021; 26(4):809. https://doi.org/10.3390/molecules26040809
Chicago/Turabian StyleUddin, Md. Zia, Arkajyoti Paul, Ahmed Rakib, Saad Ahmed Sami, Shafi Mahmud, Md. Sohel Rana, Shahadat Hossain, Abu Montakim Tareq, Mycal Dutta, Talha Bin Emran, and et al. 2021. "Chemical Profiles and Pharmacological Properties with in Silico Studies on Elatostema papillosum Wedd" Molecules 26, no. 4: 809. https://doi.org/10.3390/molecules26040809
APA StyleUddin, M. Z., Paul, A., Rakib, A., Sami, S. A., Mahmud, S., Rana, M. S., Hossain, S., Tareq, A. M., Dutta, M., Emran, T. B., & Simal-Gandara, J. (2021). Chemical Profiles and Pharmacological Properties with in Silico Studies on Elatostema papillosum Wedd. Molecules, 26(4), 809. https://doi.org/10.3390/molecules26040809