
Strategies to Target ADAM17 in Disease: From Its Discovery to the iRhom Revolution

 ,
,  and
and 
Abstract
:1. Introduction
2. Biology of ADAM17
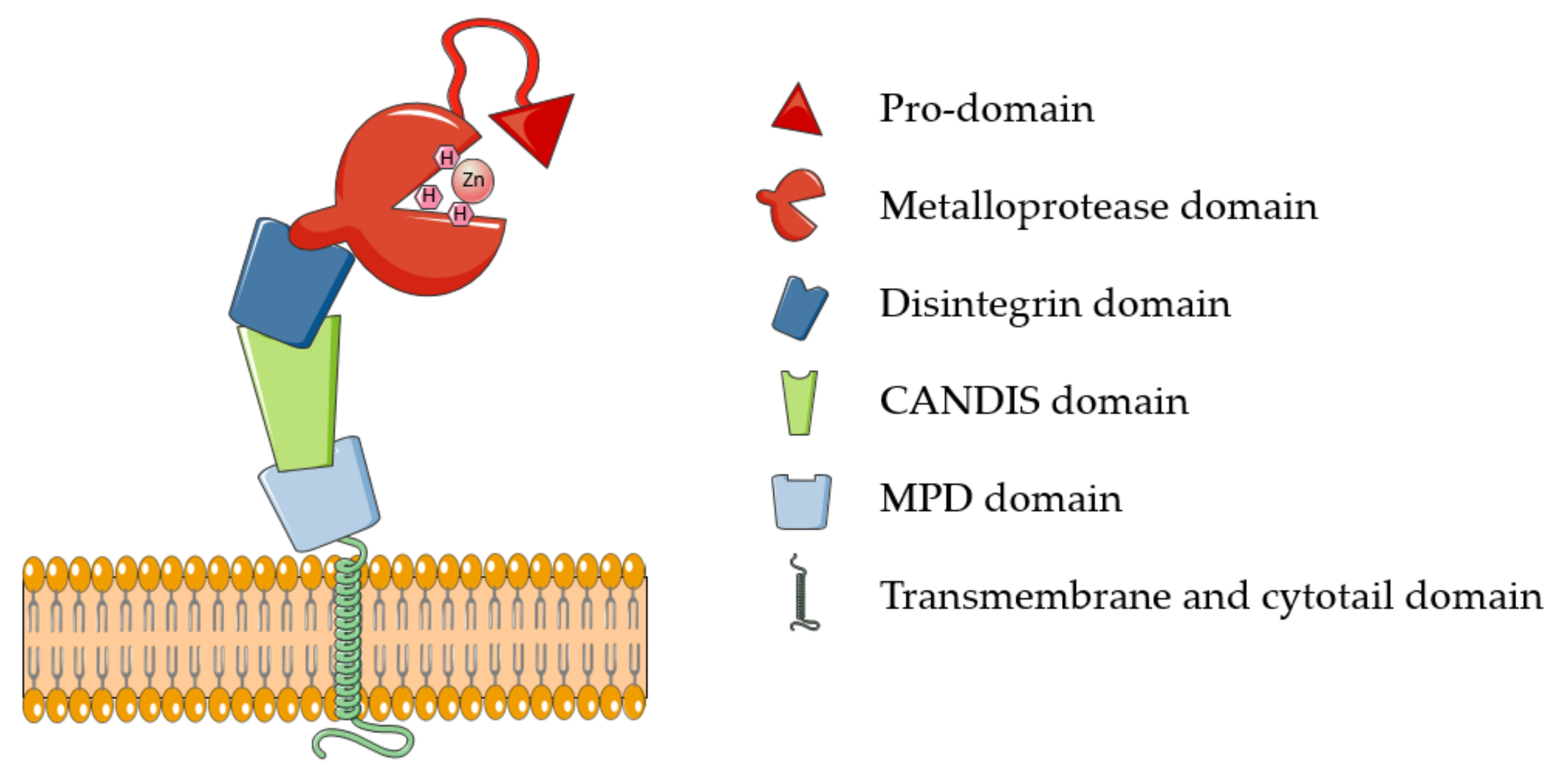
2.1. Structure
2.2. ADAM17 Function
2.3. Developmental Defects of ADAM17-Deficient Mice Are Majorly Due to Diminished EGFR Signaling
2.4. Role of ADAM17 in Inflammation
2.5. Role of ADAM17 in Skin Homeostasis
3. ADAM17 Regulation
3.1. Transcriptional and Post-transcriptional Regulation
3.2. Removal of the Pro-domain
3.3. Structural/Conformational Changes
3.3.1. Membrane Interactions
3.3.2. Protein Disulfide Isomerase
3.3.3. Integrins
3.3.4. Glycosylation
3.3.5. ADAM17 Shedding and Soluble ADAM17 Degradome
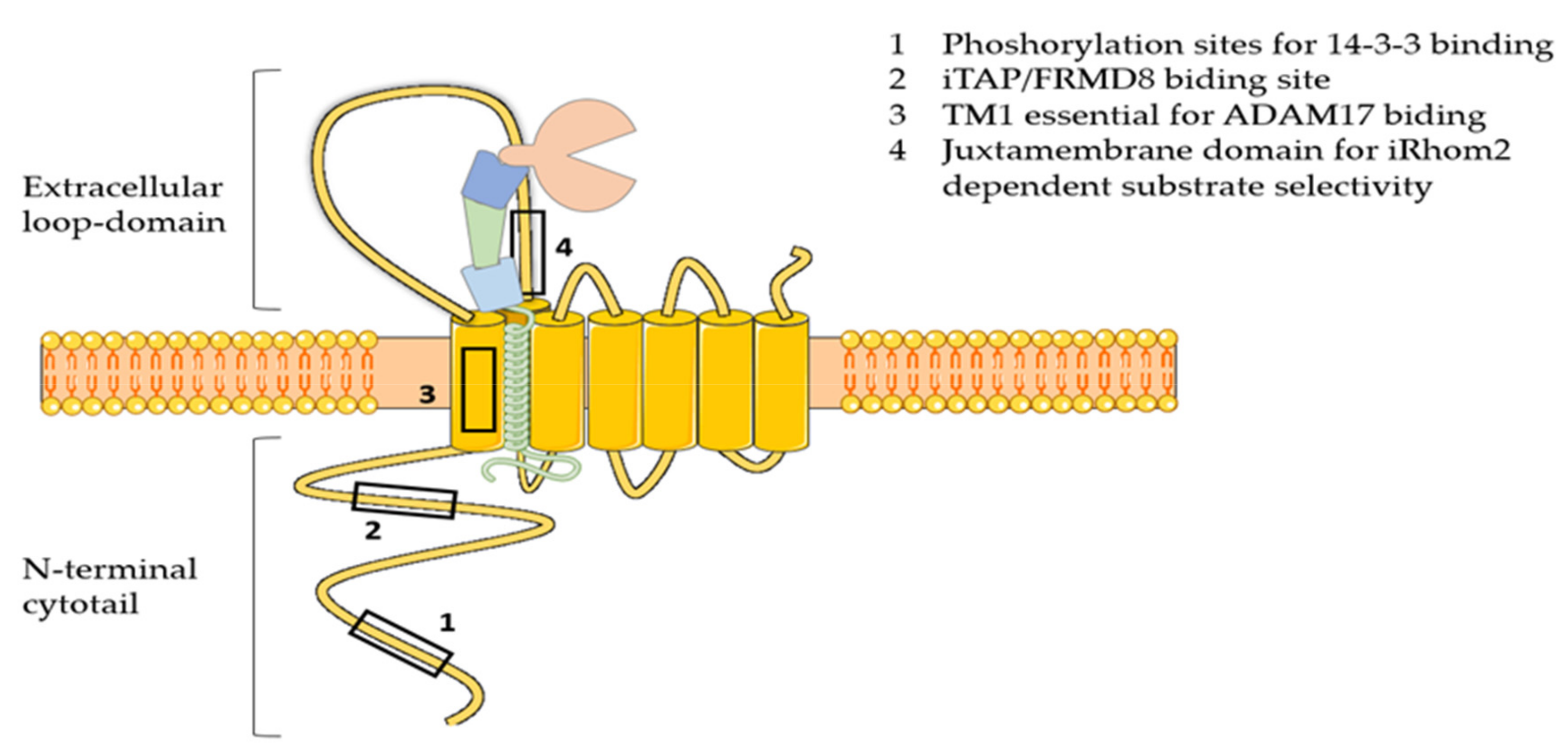
3.4. iRhoms
3.4.1. ADAM17 Trafficking and Maturation
3.4.2. ADAM17 Activation
3.4.3. TIMP-3 Inhibition
4. ADAM17 in Disease
4.1. ADAM17 in Rheumatoid Arthritis
4.2. Osteoarthritis
4.3. Lung Pathology
4.4. Atherosclerosis
4.5. Inflammatory Bowel Disease
4.6. Neurodegeneration and Alzheimer’s Disease
4.7. Nerve Recovery
4.8. ADAM17 in Cancer
4.9. COVID-19
5. Strategies for ADAM17 Inhibition
5.1. Small Molecule Inhibitors
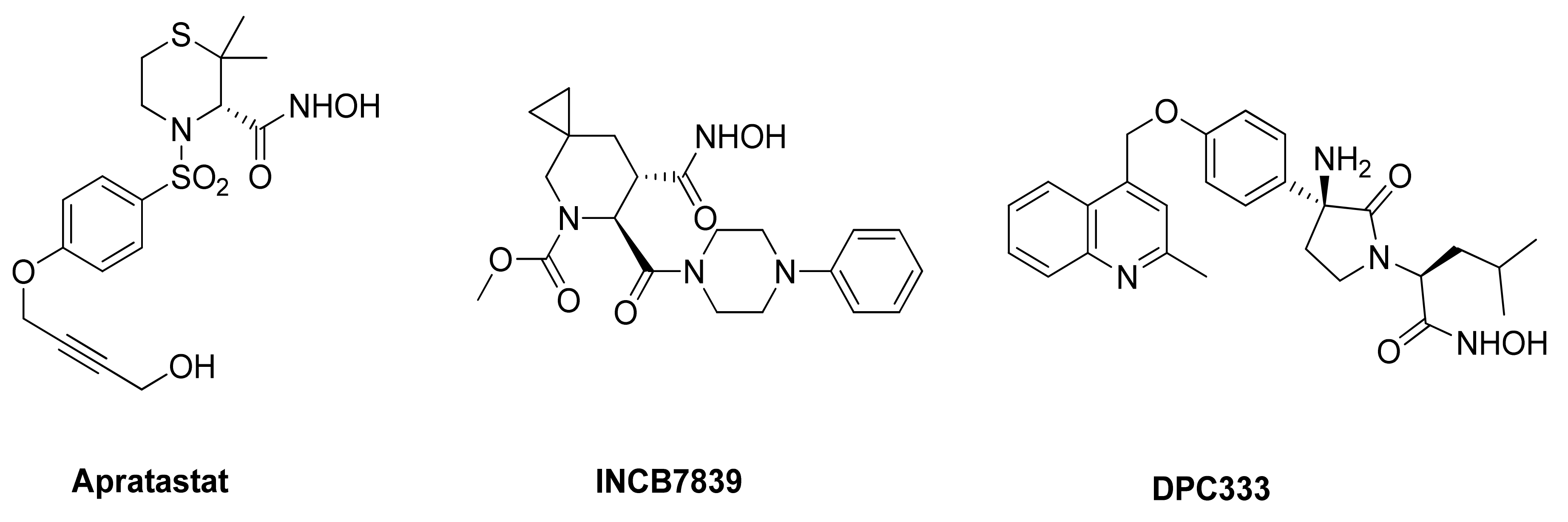
5.1.1. Hydroxamate-Based ADAM17 Inhibitors
5.1.2. Non-Hydroxamate-based ADAM17 Inhibitors
5.1.3. Non-Zinc-Binding ADAM17 Inhibitors
5.2. Pro-Domain–Dominant Negative Forms of TACE
5.3. Substrates Glycosylation
5.4. Dominant Negative Forms of ADAM17
5.5. Engineered TIMPs
5.6. Antibodies
5.7. Targeting iRhom2: Perspectives of a Revolution
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals—hardware, concepts, and recent developments. EMBO J. 2018, 37, e99456. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Feldmann, M. Anti-TNF biologic agents: Still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Murthy, A.; Khokha, R. Clipping, shedding and RIPping keep immunity on cue. Trends Immunol. 2008, 29, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane pro-tein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53. [Google Scholar] [CrossRef]
- Pennica, D.; Nedwin, G.E.; Hayflick, J.S.; Seeburg, P.H.; Derynck, R.; Palladino, M.A.; Kohr, W.J.; Aggarwal, B.B.; Goeddel, D.V. Human tumour necrosis factor: Precursor structure, expression and homology to lymphotoxin. Nat. Cell Biol. 1984, 312, 724–729. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tu-mour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.L.C.; Milla, M.E.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; Hoffman, C.E.; et al. Cloning of a disintegrin metalloproteinase that process-es precursor tumour-necrosis factor-α. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Rossello, A.; Nuti, E.; Ferrini, S.; Fabbi, M. Targeting ADAM17 Sheddase Activity in Cancer. Curr. Drug Targets 2016, 17, 1908–1927. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Hsia, H.E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srour, N.; Lebel, A.; McMahon, S.; Fournier, I.; Fugère, M.; Day, R.; Dubois, C.M. TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 2003, 554, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Chicky, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metallopro-tease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzke, C.-W.; Cobzaru, C.; Triantafyllopoulou, A.; Löffek, S.; Horiuchi, K.; Threadgill, D.W.; Kurz, T.; Van Rooijen, N.; Bruckner-Tuderman, L.; Blobel, C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Cell Biol. 2012, 197. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.C.; Hill, D.; Otero, M.; Plumb, D.A.; Froemel, D.; Dragomir, C.L.; Maretzky, T.; Boskey, A.; Crawford, H.C.; Selleri, L.; et al. ADAM17 Controls Endochondral Ossification by Regu-lating Terminal Differentiation of Chondrocytes. Mol. Cell. Biol. 2013, 33, 3077–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Miyamoto, K.; Akiyama, H.; Takaishi, H.; Morioka, H.; Nakamura, T.; Okada, Y.; Blobel, C.P.; et al. Conditional Inactivation of TACE by a Sox9 Promoter Leads to Osteoporosis and Increased Granulopoiesis via Dysregulation of IL-17 and G-CSF. J. Immunol. 2009, 182, 2093–2101. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting Edge: TNF-α-Converting Enzyme (TACE/ADAM17) Inactivation in Mouse Myeloid Cells Prevents Lethality from Endotoxin Shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Brazzell, J.; Herrera, A.H.; Walcheck, B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood 2006, 108, 2275–2279. [Google Scholar] [CrossRef]
- Bell, J.H.; Herrera, A.H.; Li, Y.; Walcheck, B. Role of ADAM17 in the ectodomain shedding of TNF- and its receptors by neutro-phils and macrophages. J. Leukoc. Biol. 2007, 82, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Wang, Y.; Herrera, A.H.; Horiuchi, K.; Walcheck, B. In Vivo role of leukocyte ADAM17 in the inflammatory and host re-sponses during E. coli -mediated peritonitis. J. Leukoc. Biol. 2010, 87, 1097–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose-John, S.; Winthrop, K.; Calabrese, L. The role of IL-6 in host defence against infections: Immunobiology and clinical impli-cations. Nat. Rev. Rheumatol. 2017, 13, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway1. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, L.H.; Rose-John, S. IL-6 biology: Implications for clinical targeting in rheumatic disease. Nat. Rev. Rheumatol. 2014, 10, 720–727. [Google Scholar] [CrossRef]
- Yan, I.; Schwarz, J.; Luecke, K.; Schumacher, N.; Schumacher, V.; Schmidt, S.; Rabe, B.; Saftig, P.; Donners, M.M.P.C.; Rose-John, S.; et al. ADAM17 controls IL-6 signaling by cleavage of the murine IL-6Rα from the cell surface of leukocytes during inflammatory responses. J. Leukoc. Biol. 2015, 99, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Schütze, S.; Hartmann, D.; Matthews, V.; Schuster, B.; Bussmeyer, I.; Ludwig, A.; Hundhausen, C.; Sadowski, T.; Saftig, P.; Kallen, K.-J.; et al. Cellular Cholesterol Depletion Triggers Shedding of the Human Interleukin-6 Receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 2003, 278, 38829–38839. [Google Scholar] [CrossRef] [Green Version]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340. [Google Scholar] [CrossRef]
- Murthy, A.; Shao, Y.W.; Narala, S.R.; Molyneux, S.D.; Zúñiga-Pflücker, J.C.; Khokha, R. Notch Activation by the Metalloproteinase ADAM17 Regulates Myeloproliferation and Atopic Barrier Immunity by Suppressing Epithelial Cytokine Synthesis. Immunity 2012, 36, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, L.M. The ADAM family insights into Notch proteolysis. Fly 2012, 6, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duojia, P.; Rubin, G.M. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 1997, 90, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Gasson, J.C. Molecular physiology of granulocyte-macrophage colonystimulating factor. Blood 1991, 77, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Reche, P.A.; Soumelis, V.; Gorman, D.M.; Clifford, T.; Liu, M.-R.; Travis, M.; Zurawski, S.M.; Johnston, J.; Liu, Y.-J.; Spits, H.; et al. Human Thymic Stromal Lymphopoietin Preferentially Stimulates Myeloid Cells. J. Immunol. 2001, 167, 336–343. [Google Scholar] [CrossRef]
- Ebner, S.; Nguyen, V.A.; Forstner, M.; Wang, Y.H.; Wolfram, D.; Liu, Y.J.; Romani, N. Thymic stromal lymphopoietin converts human epidermal Langerhans cells into antigenpresenting cells that induce proallergic T cells. J. Allergy Clin. Immunol. 2007, 119, 982–990. [Google Scholar] [CrossRef]
- Ringel, J.; Jesnowski, R.; Moniaux, N.; Lüttges, J.; Ringel, J.; Choudhury, A.; Batra, S.K.; Kloppel, G.; Lohr, M. Aberrant expression of a disintegrin and metalloproteinase 17/tumor necrosis factor-α converting enzyme increases the malignant potential in human pancreatic ductal ade-nocarcinoma. Cancer Res. 2006, 66, 9045–9053. [Google Scholar] [CrossRef] [Green Version]
- Blanchot-Jossic, F.; Jarry, A.; Masson, D.; Bach-Ngohou, K.; Paineau, J.; Denis, M.G.; Laboisse, C.L.; Mosnier, J.-F. Up-regulated expression of ADAM17 in human colon carcinoma: Coexpression with EGFR in neoplastic and endothelial cells. J. Pathol. 2005, 207, 156–163. [Google Scholar] [CrossRef]
- Wawro, K.; Wawro, M.; Strzelecka, M.; Czarnek, M.; Bereta, J. The role of NF-κB and Elk-1 in the regulation of mouse ADAM17 expression. Biol. Open. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Chen, B.; Wu, J.; Jiang, C.; Fan, Z.; Feng, Y.; Xu, Y. Epigenetic Regulation of a Disintegrin and Metalloproteinase (ADAM) Transcription in Colorectal Cancer Cells: Involvement of β-Catenin, BRG1, and KDM4. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Doberstein, K.; Steinmeyer, N.; Hartmetz, A.K.; Eberhardt, W.; Mittelbronn, M.; Harter, P.N.; Juengel, E.; Blaheta, R.; Pfeilschifter, J.; Gutwein, P. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia US 2013, 15. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chopp, M.; Zheng, X.; Katakowski, M.; Buller, B.; Jiang, F. MiR-145 reduces ADAM17 expression and inhibits in vitro migra-tion and invasion of glioma cells. Oncol. Rep. 2013, 29, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.W.; Zhang, L.J.; Huang, X.H.; Chen, L.Z.; Su, Q.; Zeng, W.T.; Li, W.; Wang, Q. MiR-145 suppresses cell invasion in hepato-cellular carcinoma cells: MiR-145 targets ADAM17. Hepatol. Res. 2014, 44, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, Q.; Gui, H.; Xu, D.-P.; Yang, Y.-L.; Su, D.-F.; Liu, X. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 2013, 23, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Wang, Z.; Zhang, J.; Zhou, H.; Jin, L.; Bai, R.; Weng, Y. Adam17, a Target of Mir-326, Promotes Emt-Induced Cells Invasion in Lung Adenocarcinoma. Cell. Physiol. Biochem. 2015, 36, 1175–1185. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Y.; Zhou, H.; Lei, L.; Xu, L. MicroRNA-152 targets ADAM17 to suppress NSCLC progression. FEBS Lett. 2014, 588, 1983–1988. [Google Scholar] [CrossRef]
- Buckley, C.A.; Rouhani, F.N.; Kaler, M.; Adamik, B.; Hawari, F.I.; Levine, S.J. Amino-terminal TACE prodomain attenuates TNFR2 cleavage independently of the cysteine switch. Am. J. Physiol. Cell. Mol. Physiol. 2005, 288, 1132–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.; Maretzky, T.; Peleg, Y.; Blobel, C.P.; Sagi, I. The Functional Maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 Requires Processing at a Newly Identified Proprotein Convertase (PC) Cleavage Site. J. Biol. Chem. 2015, 290, 12135–12146. [Google Scholar] [CrossRef] [Green Version]
- Pavlenko, E.; Cabron, A.-S.; Arnold, P.; Dobert, J.P.; Rose-John, S.; Zunke, F. Functional Characterization of Colon Cancer-Associated Mutations in ADAM17: Modifications in the Pro-Domain Interfere with Trafficking and Maturation. Int. J. Mol. Sci. 2019, 20, 2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellier, E.; Canault, M.; Poggi, M.; Bonardo, B.; Nicolay, A.; Alessi, M.-C.; Nalbone, G.; Peiretti, F. HDLs activate ADAM17-dependent shedding. J. Cell. Physiol. 2007, 214, 687–693. [Google Scholar] [CrossRef]
- Tellier, E.; Canault, M.; Rebsomen, L.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. The shedding activity of ADAM17 is seques-tered in lipid rafts. Exp. Cell Res. 2006, 312. [Google Scholar] [CrossRef]
- Sommer, A.; Kordowski, F.; Büch, J.; Maretzky, T.; Evers, A.; Andrä, J.; Düsterhöft, S.; Michalek, M.; Lorenzen, I.; Somasundaram, P.; et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 2016, 7, 11523. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, M.C.; Wagner-Britz, L.; Nguyen, D.B.; Asanidze, S.; Mutua, J.; Mohamed, N.; Hanf, B.; Ghashghaeinia, M.; Kaestner, L.; Bernhardt, I. Novel Insights in the Regulation of Phosphatidylserine Exposure in Human Red Blood Cells. Cell. Physiol. Biochem. 2016, 39, 1941–1954. [Google Scholar] [CrossRef]
- Veit, M.; Ahrens, B.; Seidel, J.; Sommer, A.; Bhakdi, S.; Reiss, K. Mutagenesis of the ADAM17-phosphatidylserine–binding motif leads to embryonic lethality in mice. Life Sci. Alliance 2019, 2, e201900430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willems, S.H.; Tape, C.J.; Stanley, P.L.; Taylor, N.A.; Mills, I.G.; Neal, D.E.; Mc Cafferty, J.; Murphu, G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem. J. 2010, 428. [Google Scholar] [CrossRef] [Green Version]
- Bax, D.V.; Messent, A.J.; Tart, J.; Hoang, M.V.; Kott, J.; Maciewicz, R.A.; Humphries, N.J. Integrin α5β1 and ADAM-17 interact In Vitro and colocalize in migrating HeLa cells. J. Biol. Chem. 2004, 279. [Google Scholar] [CrossRef] [Green Version]
- Chavaroche, A.; Cudic, M.; Giulianotti, M.; Houghten, R.A.; Fields, G.B.; Minond, D. Glycosylation of a disintegrin and metaloprotease 17 affects its activity and inhibition. Anal. Biochem. 2014, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharfenberg, F.; Helbig, A.; Sammel, M.; Benzel, J.; Schlomann, U.; Peters, F.; Wichert, R.; Bettendorff, M.; Schmidt-Arras, D.; Rose-John, S.; et al. Degradome of soluble ADAM10 and ADAM17 metalloproteases. Cell. Mol. Life Sci. 2019, 77, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Groth, E.; Pruessmeyer, J.; Babendreyer, A.; Schumacher, J.; Pasqualon, T.; Dreymueller, D.; Higashiyama, S.; Lorenzen, I.; Grötzinger, J.; Cataldo, D.; et al. Stimulated release and functional activity of surface expressed metalloproteinase ADAM17 in exosomes. Biochim. Biophys. Acta (BBA)Bioenerg. 2016, 1863, 2795–2808. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Kesti, T.; Uğurlu, H.; Baur, A.S.; Fagerlund, R.; Saksela, K. Tyrosine phosphorylation directs TACE into extracellular vesi-cles via unconventional secretion. Traffic 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Dulloo, I.; Muliyil, S.; Freeman, M. The molecular, cellular and pathophysiological roles of iRhom pseudoproteases. Open Biol. 2019, 9, 190003. [Google Scholar] [CrossRef] [Green Version]
- Weskamp, G.; Tüshaus, J.; Li, D.; Feederle, R.; Maretzky, T.; Swendemann, S.; Pedersen, E.F.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. ADAM17 stabilizes its interacting partner inactive Rhomboid 2 (iRhom2) but not inactive Rhomboid 1 (iRhom1). J. Biol. Chem. 2020, 295. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 Regulation of TACE Controls TNF-Mediated Protection Against Listeria and Responses to LPS. Science 2012, 335, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote traficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christova, Y.; Adrain, C.; Bambrough, P.; Ibrahim, A.; Freeman, M. Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 2013, 14, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Maretzky, T.; Weskamp, G.; Monette, S.; Qing, X.; Issuree, P.D.A.; Crawford, H.C.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 6080–6085. [Google Scholar] [CrossRef] [Green Version]
- Hosur, V.; Low, B.E.; Li, D.; Stafford, G.A.; Kohar, V.; Shultz, L.D.; Wiles, M.V. Genes adapt to outsmart gene-targeting strategies in mutant mouse strains by skipping exons to reinitiate transcription and translation. Genome Biol. 2020, 21, 1–25. [Google Scholar] [CrossRef]
- Arora, P.S.; Cuevas, B.D.; Russo, A.; Johnson, G.L.; Trejo, J. Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 2008, 27, 4434–4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, R.R.; Bair, J.A.; Carozza, R.B.; Li, D.; Shatos, M.A.; Dartt, D.A. Signaling pathways used by EGF to stimulate conjunctival goblet cell secretion. Exp. Eye Res. 2012, 103. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.A.; Niu, X.D.; Reiss, K.; Saftig, P.; Khokha, P.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mech-anism that controls access to its catalytic site. J. Cell Sci. 2010, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Rodríguez, E.; Montero, J.C.; Esparís-Ogando, A.; Yuste, L.; Pandiella, A.; Lampugnani, M.G.; Zanetti, A.; Breviario, F.; Balconi, G.; Orsenigo, F.; et al. Extracellular Signal-regulated Kinase Phosphorylates Tumor Necrosis Factor α-converting Enzyme at Threonine 735: A Potential Role in Regulated Shedding. Mol. Biol. Cell 2002, 13, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Elliott, K.J.; Bourne, A.M.; Takayanagi, T.; Takaguri, A.; Kobayashi, T.; Eguchi, K.; Eguchi, S. ADAM17 silencing by adenovirus encoding miRNA-embedded siRNA revealed essential signal transduction by angiotensin II in vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2013, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, K.C.; Blobel, C.P. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phos-phorylation at threonine 735. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; Monette, S.; Weskamp, G.; le Gall, S.; Beutler, B.; Weinstein, H.; Blobel, C.P. Structural modeling defines transmembrane resi-dues in ADAM17 that are crucial for Rhbdf2-ADAM17-dependent proteolysis. J. Cell Sci. 2017, 130. [Google Scholar] [CrossRef] [Green Version]
- Cavadas, M.; Oikonomidi, I.; Gaspar, C.J.; Burbridge, E.; Badenes, M.; Félix, I.; Bolado, A.; Hu, T.; Bileck, A.; Gerner, C.; et al. Phosphorylation of iRhom2 Controls Stimulated Proteolytic Shedding by the Metalloprotease ADAM17/TACE. Cell Rep. 2017, 21. [Google Scholar] [CrossRef] [Green Version]
- Grieve, A.G.; Xu, H.; Künzel, U.; Bambrough, P.; Sieber, B.; Freeman, M. Phosphorylation of iRhom2 at the plasma membrane con-trols mammalian TACE-dependent inflammatory and growth factor signalling. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Oikonomidi, I.; Burbridge, E.; Cavadas, M.; Sullivan, G.; Collis, B.; Naegele, H.; Clancy, D.; Brezinova, J.; Hu, T.; Bileck, A.; et al. iTAP, a novel iRhom interactor, controls TNF secretion by policing the stability of iRhom/TACE. eLife 2018, 7, e35032. [Google Scholar] [CrossRef] [PubMed]
- Künzel, U.; Grieve, A.G.; Meng, Y.; Sieber, B.; Cowley, S.A.; Freeman, M. FRMD8 promotes inflammatory and growth factor signal-ling by stabilising the iRhom/ADAM17 sheddase complex. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.A.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; McIlwain, D.; Xie, Y.; Zheng, Y.; Mak, T.W.; Weinstein, H.; Blobel, C.P. Substrate-selective protein ectodomain shedding by ADAM17 and iRhom2 depends on their juxtamembrane and transmembrane domains. FASEB J. 2020, 34, 4956–4969. [Google Scholar] [CrossRef] [Green Version]
- Iwagishi, R.; Tanaka, R.; Seto, M.; Takagi, T.; Norioka, N.; Ueyama, T.; Kawamura, T.; Takagi, J.; Ogawa, Y.; Shirakabe, K. Negatively charged amino acids in the stalk region of membrane proteins reduce ectodomain shedding. J. Biol. Chem. 2020, 295, 12343–12352. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional di-versity. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2010, 1803, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Wisniewska, M.; Goettig, P.; Maskos, K.; Belouski, E.; Winters, D.; Hecht, R.; Black, R.; Bode, W. Structural Determinants of the ADAM Inhibition by TIMP-3: Crystal Structure of the TACE-N-TIMP-3 Complex. J. Mol. Biol. 2008, 381, 1307–1319. [Google Scholar] [CrossRef]
- Mahmoodi, M.; Sahebjam, S.; Smookler, D.; Khokha, R.; Mort, J.S. Lack of tissue inhibitor of metalloproteinases-3 results in an en-hanced inflammatory response in antigen-induced arthritis. Am. J. Pathol. 2005, 166. [Google Scholar] [CrossRef] [Green Version]
- Sahebjam, S.; Khokha, R.; Mort, J.S. Increased collagen and aggrecan degradation with age in the joints of Timp3(-/-) mice. Ar-thritis Rheum. 2007, 56, 905–909. [Google Scholar] [CrossRef]
- Black, R.A.; Slack, J.; Tocker, J.; Eisenman, J.; Jacobson, E.; Delaney, J.; Winters, D.; Hecht, R.; Bendele, A. A14 injected TIMP-3 protects cartilage in a rat meniscal tear model. Osteoarthr. Cartil. 2006, 14. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, F.F.; Smookler, D.S.; Taylor, S.E.; Fingleton, B.; Kassiri, Z.; Sanchez, O.H.; English, J.L.; Matrisian, L.M.; Au, B.; Yeh, W.C.; et al. Abnormal TNF activity in Timp3-/- mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat. Genet. 2004, 36, 969–977. [Google Scholar] [CrossRef]
- Federici, M.; Hribal, M.L.; Menghini, R.; Kanno, H.; Marchetti, V.; Porzio, O.; Sunnarborg, S.W.; Rizza, S.; Serino, M.; Cunsolo, V.; et al. Timp3 deficiency in insulin receptor-haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF-. J. Clin. Investig. 2005, 115, 3494–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casagrande, V.; Mauriello, A.; Bischetti, S.; Mavilio, M.; Federici, M.; Menghini, R. Hepatocyte specific TIMP3 expression prevents diet dependent fatty liver disease and hepatocellular carcinoma. Sci. Rep. 2017, 7, 6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menghini, R.; Casagrande, V.; Menini, S.; Marino, A.; Marzano, V.; Hribal, M.L.; Gentileschi, P.; Lauro, D.; Schillaci, O.; Pugliese, G.; et al. TIMP3 overexpression in macrophages protects from insulin resistance, adipose inflammation, and nonalcoholic fatty liver disease in mice. Diabetes 2012, 61. [Google Scholar] [CrossRef] [Green Version]
- Casagrande, V.; Menghini, R.; Menini, S.; Marino, A.; Marchetti, V.; Cavalera, M.; Fabrizi, M.; Hribal, M.L.; Pugliesse, G.; Gentileschi, P.; et al. Overexpression of tissue inhibitor of metalloproteinase 3 in macrophages reduces atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Bondeson, J.; Feldmann, M.; Maini, R.N. TNF as a therapeutic target. Immunologist 2000, 8, 136–140. [Google Scholar]
- Cope, A.P.; Gibbons, D.; Brennan, F.M.; Feldmann, M.; Aderka, D.; Doherty, M.; Jones, A.C.; Engelmann, H.L.; Wallach, D.; Maini, R.N. Increased levels of soluble tumor necrosis fac-tor receptors in the sera and synovial fluid of patients with rheumatic diseases. Arthritis Rheum. 1992, 35. [Google Scholar] [CrossRef]
- Brennan, F.M.; Jackson, A.; Chantry, D.; Maini, R.; Feldmann, M. Inhibitory effect of tnfα antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet 1989, 2, 244–247. [Google Scholar] [CrossRef]
- Issuree, P.D.; Maretzky, T.; McIlwain, D.R.; Monette, S.; Qing, X.; Lang, P.A.; Swendeman, S.L.; Park-Min, K.-H.; Binder, N.; Kalliolias, G.D.; et al. iRHOM2 is a critical pathogenic mediator of in-flammatory arthritis. J. Clin. Invest. 2013, 123, 928–932. [Google Scholar] [PubMed] [Green Version]
- Alexopoulou, L.; Kranidioti, K.; Xanthoulea, S.; Denis, M.; Kotanidou, A.; Douni, E.; Blackshear, P.J.; Kontoyiannis, D.L.; Kollias, G. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur. J. Immunol. 2006, 36, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Secher, T.; Ezhevsky, S.A.; Janot, L.; Steed, P.M.; O’Brien, C.; Eivazi, A.; Kung, J.; Nguyen, D.-H.T. Dominant-Negative Inhibitors of Soluble TNF Atten-uate Experimental Arthritis without Suppressing Innate Immunity to Infection. J. Immunol. 2007, 179. [Google Scholar] [CrossRef] [Green Version]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Grell, M.; Wajant, H.; Zimmermann, G.; Scheurich, P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1998, 95, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H.; Scheurich, P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 2011, 278. [Google Scholar] [CrossRef]
- Chen, X.; Wu, X.; Zhou, Q.; Howard, O.M.Z.; Netea, M.G.; Oppenheim, J.J. TNFR2 Is Critical for the Stabilization of the CD4+Foxp3+ Regulatory T Cell Phenotype in the Inflammatory Environment. J. Immunol. 2013, 190, 1076–1084. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, N.; Rose-John, S. ADAM17 Activity and IL-6 Trans-Signaling in Inflammation and Cancer. Cancers 2019, 11, 1736. [Google Scholar] [CrossRef] [Green Version]
- Nowell, M.A.; Williams, A.S.; Carty, S.A.; Scheller, J.; Hayes, A.J.; Jones, G.W.; Richards, P.J.; Slinn, S.; Ernst, M.; Jenkins, B.J.; et al. Therapeutic Targeting of IL-6 Trans Signaling Counteracts STAT3 Control of Experimental Inflammatory Arthritis. J. Immunol. 2008, 182, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, A.; Hirano, T.; Hishitani, Y.; Tanaka, T. Safety and Efficacy of Tocilizumab for the Treatment of Rheumatoid Arthritis. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2012, 5, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Ogata, A.; Kato, Y.; Higa, S.; Yoshizaki, K. IL-6 inhibitor for the treatment of rheumatoid arthritis: A comprehensive review. Mod. Rheumatol. 2019, 29, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.-P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2010, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Saklatvala, J. Tumour necrosis factor α stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nat. Cell Biol. 1986, 322, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Peeters-Joris, C.; Vaes, G. Modulation by interleukin 1 and tumor necrosis factor α of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochim. Biophys. Acta (BBA) Bioenerg. 1990, 1052, 366–378. [Google Scholar] [CrossRef]
- Reboul, P.; Pelletier, J.P.; Tardif, G.; Cloutier, J.M.; Martel-Pelletier, J. The new collagenase, collagenase-3, is expressed and synthe-sized by human chondrocytes but not by synoviocytes: A role in osteoarthritis. J. Clin. Investig. 1996, 97. [Google Scholar] [CrossRef]
- Philp, A.M.; Davis, E.T.; Jones, S.W. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology 2016, 56, 278–881. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, J.; Tran, H.; Verbeek, M.M.; Reiss, K.; Estus, S.; Bu, G. LRP1 shedding in human brain: Roles of ADAM10 and AD-AM17. Mol. Neurodegener. 2009, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Troeberg, L.; Scilabra, S.D.; Pelosi, M.; Murphy, C.L.; Strickland, D.K.; Nagase, H. LRP-1-mediated endocytosis regulates ex-tracellular activity of ADAMTS-5 in articular cartilage. FASEB J. 2013, 27. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Owen, K.; Parker, A.E.; Scilabra, S.D.; Dudhia, J.; Strickland, D.K.; Troeberg, L.; Nagase, H. Low density lipoprotein receptor-related pro-tein 1 (LRP1)-mediated endocytic clearance of a disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4): Functional differences of non-catalytic domains of ADAMTS-4 and ADAMTS-5 in LRP1 binding. J. Biol. Chem. 2014, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Okano, H.; Miyagawa, W.; Visse, R.; Shitomi, Y.; Santamaria, S.; Dudhia, J.; Troeberg, L.; Strickland, D.K.; Hirohata, S.; et al. MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 2016, 56, 57–73. [Google Scholar] [CrossRef]
- Carreca, A.P.; Pravatà, V.M.; Markham, M.; Bonelli, S.; Murphy, G.; Nagase, H.; Troeberg, L.; Scilabra, S.D. TIMP-3 facilitates binding of target metallopro-teinases to the endocytic receptor LRP-1 and promotes scavenging of MMP-1. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Scilabra, S.D.; Pigoni, M.; Pravatá, V.; Schätzl, T.; Müller, S.A.; Troeberg, L.; Lichtenthaler, S.F. Increased TIMP-3 expression alters the cellular se-cretome through dual inhibition of the metalloprotease ADAM10 and ligand-binding of the LRP-1 receptor. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Santamaria, S.; Botkjaer, K.A.; Dudhia, J.; Troeberg, L.; Itoh, Y.; Murphy, G.; Nagase, H. Inhibition of Shedding of Low-Density Lipopro-tein Receptor-Related Protein 1 Reverses Cartilage Matrix Degradation in Osteoarthritis. Arthritis Rheumatol. 2017, 69, 1246–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorovoy, M.; Gaultier, A.; Campana, W.M.; Firestein, G.S.; Gonias, S.L. Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. J. Leukoc. Biol. 2010, 88, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Arndt, P.G.; Strahan, B.; Wang, Y.; Long, C.; Horiuchi, K.; Walcheck, B. Leukocyte adam17 regulates acute pulmonary inflammation. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Dreymueller, D.; Martin, C.; Kogel, T.; Pruessmeyer, J.; Hess, F.M.; Horiuchi, K.; Uhlig, S.; Ludwig, A. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol. Med. 2012, 4, 412–423. [Google Scholar] [CrossRef]
- Shiomi, T.; Tschumperlin, D.J.; Park, J.-A.; Sunnarborg, S.W.; Horiuchi, K.; Blobel, C.P.; Drazen, J.M. TNF-α–Converting Enzyme/A Disintegrin and Metalloprotease−17 Mediates Mechanotransduction in Murine Tracheal Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Stolarczyk, M.; Scholte, B.J. The EGFR-ADAM17 Axis in Chronic Obstructive Pulmonary Disease and Cystic Fibrosis Lung Pathology. Mediat. Inflamm. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaou, A.; Zhao, Z.; Northoff, B.H.; Sass, K.; Herbst, A.; Kohlmaier, A.; Chalaris, A.; Wolfrum, C.; Weber, C.; Steffens, S.; et al. Adam17 Deficiency promotes atherosclerosis by en-hanced TNFR2 signaling in Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKellar, G.E.; McCarey, D.W.; Sattar, N.; McInnes, I.B. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat. Rev. Cardiol. 2009, 6, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Reinecker, H.C.; Steffen, M.; Witthoeft, T.; Pflueger, I.; Schreiber, S.; MacDermott, R.P.; Raedler, A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1β by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin. Exp. Immunol. 1993, 94. [Google Scholar] [CrossRef]
- Murch, S.H.; Braegger, C.P.; Walker-Smith, J.A.; Macdonald, T.T. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut 1993, 34, 1705–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berns, M.; Hommes, D.W. Anti-TNF-α therapies for the treatment of Crohn’s disease: The past, present and future. Expert Opin. Investig. Drugs 2016, 25, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Horiuchi, K.; Sasaki, A.; Tsukamoto, T.; Okabayashi, K.; Hasegawa, H.; Kitagawa, Y.; Okada, Y. Epithelial Cell-Derived a Disintegrin and Metalloproteinase-17 Confers Resistance to Colonic Inflammation Through EGFR Activation. EBioMedicine 2016, 5, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaydon, D.C.; Biancheri, P.; Di, W.-L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; Van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory Skin and Bowel Disease Linked toADAM17Deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Geesala, R.; Schanz, W.; Biggs, M.; Dixit, G.; Skurski, J.; Gurung, P.; Mayerholz, D.K.; Elliott, D.; Issuree, P.D.; Maretzky, T. Loss of RHBDF2 results in an early-onset spontaneous mu-rine colitis. J. Leukoc. Biol. 2019, 105. [Google Scholar] [CrossRef]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.T.; Collins, J.S.; Wiener, H.; Acton, R.; Go, R.C.P. The role of TNF and its receptors in Alzheimer’s disease. Neurobiol. Aging 2001, 22. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against is-chemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P.Y. TNFα promotes proliferation of oligodendrocyte pro-genitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef]
- Giuliani, F.; Vernay, A.; Leuba, G.; Schenk, F. Decreased behavioral impairments in an Alzheimer mice model by interfering with TNF-alpha metabolism. Brain Res. Bull. 2009, 80, 302–308. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amy-loid β generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18. [Google Scholar] [CrossRef] [Green Version]
- Naude, P.J.W.; Dobos, N.; van der Meer, D.; Mulder, C.; Pawironadi, K.G.D.; den Boer, J.A.; van der Zee, E.A.; Luiten, P.G.M.; Eisel, U.L.M. Analysis of cognition, motor perfor-mance and anxiety in young and aged tumor necrosis factor alpha receptor 1 and 2 deficient mice. Behav. Brain Res. 2014, 258. [Google Scholar] [CrossRef]
- Montgomery, S.L.; Mastrangelo, M.A.; Habib, D.; Narrow, W.C.; Knowlden, S.A.; Wright, T.W.; Bowers, W.J. Ablation of TNF-RI/RII expres-sion in Alzheimer’s disease mice leads to an unexpected enhancement of pathology: Implications for chronic pan-TNF-α sup-pressive therapeutic strategies in the brain. Am. J. Pathol. 2011, 179. [Google Scholar] [CrossRef]
- Shi, J.-Q.; Wang, B.-R.; Jiang, W.-W.; Chen, J.; Zhu, Y.-W.; Zhong, L.-L.; Zhang, Y.-D.; Xu, J. Cognitive improvement with intrathecal administration of infliximab in a woman with Alzheimer’s disease. J. Am. Geriatr. Soc. 2011, 59, 1142–1144. [Google Scholar] [CrossRef]
- McAlpine, F.E.; Lee, J.-K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rosner, S.; Lichtenthaler, S. ADAM10 is the physiologically relevant, constitu-tive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29. [Google Scholar] [CrossRef] [Green Version]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Hass, K.; Fahrenholz, F. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, D.; May, P.; Gu, W.; Mayhaus, M.; Pichler, S.; Spaniol, C.; Glaab, E.; Bobbili, D.R.; Antony, P.; Koegelsberger, S.; et al. A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol. Psychiatry 2018, 25, 629–639. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; O’Hara, B.F.; Blobel, C.P. iRhoms in the brain—A new frontier? Cell Cycle 2015, 14, 3003–3004. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Sommer, D.; Corstjens, I.; Sanchez, S.; Dooley, D.; Lemmens, S.; Van Broeckhoven, J.; Bogie, J.; Vanmierlo, T.; Vidal, P.M.; Rose-John, S.; et al. ADAM17-deficiency on microglia but not on macrophages promotes phagocytosis and functional recovery after spinal cord injury. Brain Behav. Immun. 2019, 80, 129–145. [Google Scholar] [CrossRef]
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trumbach, D.; Wurts, W.; et al. TREM 2 deficiency impairs chemotaxis and mi-croglial responses to neuronal injury. EMBO Rep. 2017, 18, 1186–1198. [Google Scholar] [CrossRef]
- Feuerbach, D.; Schindler, P.; Barske, C.; Joller, S.; Beng-Louka, E.; Worringer, K.A.; Kommineni, S.; Kaykas, A.; Ho, D.J.; Ye, C.; et al. ADAM17 is the main sheddase for the gen-eration of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci. Lett. 2017, 660. [Google Scholar] [CrossRef]
- Tanaka, Y.; Miyamoto, S.; Suzuki, S.O.; Oki, E.; Yagi, H.; Sonoda, K.; Yamazaki, A.; Mizushima, H.; Maehara, Y.; Mekada, E.; et al. Clinical Significance of Heparin-Binding Epidermal Growth Factor-Like Growth Factor and A Disintegrin and Metalloprotease 17 Expression in Human Ovarian Cancer. Clin. Cancer Res. 2005, 11, 4783–4792. [Google Scholar] [CrossRef] [Green Version]
- Borrell-Pagès, M.; Rojo, F.; Albanell, J.; Baselga, J.; Arribas, J. TACE is required for the activation of the EGFR by TGF-α in tumors. EMBO J. 2003, 22, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knösel, T.; et al. ADAM17 is required for EGF-R–induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225. [Google Scholar] [CrossRef] [PubMed]
- Dosch, J.; Ziemke, E.; Wan, S.; Luker, K.; Welling, T.; Hardiman, K.; Fearon, E.; Thomas, S.; Flynn, M.; Rios-Doria, J.; et al. Targeting ADAM17 inhibits human colorectal adenocarci-noma progression and tumor-initiating cell frequency. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Czarnek, M.; Bzowska, M.; Mezyk-Kopeć, R.; Stalińska, K.; Wyroba, B.; Sroka, J.; Jucha, J.; Deneka, D.; Stoklova, P.; et al. ADAM17 Silencing in Mouse Colon Carcino-ma Cells: The Effect on Tumoricidal Cytokines and Angiogenesis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Giricz, O.; Calvo, V.; Peterson, E.A.; Abouzeid, C.M.; Kenny, P.A. TACE-dependent TGFα shedding drives triple-negative breast cancer cell invasion. Int. J. Cancer 2013, 133, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Q.; Kim, B.G.; Kang, S.; Choi, Y.P.; Yoon, J.H.; Cho, N.H. Human breast cancer-associated fibroblasts enhance cancer cell pro-liferation through increased TGF-α cleavage by ADAM17. Cancer Lett. 2013, 336. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; McGowan, P.M.; Mullooly, M.; Murray, A.; Synnott, N.; O’Donovan, N.; Flanagan, L.; Tape, C.J.; Murphy, G.; Crown, J.; et al. Targeting ADAM-17 with an inhibitory monoclonal antibody has antitumour effects in triple-negative breast cancer cells. Br. J. Cancer 2015, 112, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Weskamp, G.; Mendelson, K.; Swendeman, S.; le Gall, S.; Ma, Y.; Lyman, S.; Hinoki, A.; Eguchi, S.; Guaiquil, V.; Horiuchi, K.; et al. Pathological neovascularization is reduced by in-activation of ADAM17 in endothelial cells but not in pericytes. Circ. Res. 2010, 106, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, Y.; Wise, R.; Zolkiewska, A. Proteolytic processing of PD-L1 by ADAM proteases in breast cancer cells. Cancer Immunol. Immunother. 2020, 69, 43–55. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Orme, J.J.; Jazieh, K.A.; Xie, T.; Harrington, S.; Liu, X.; Ball, M.; Madden, B.; Charlesworth, M.C.; Azam, T.U.; Lucien, F.; et al. ADAM10 and ADAM17 cleave PD-L1 to mediate PD-(L)1 in-hibitor resistance. OncoImmunology 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Newton, R.; Bahaie, N.S.; Long, C.; Walcheck, B. ADAM17 cleaves CD16b (FcγRIIIb) in human neutrophils. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1833, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181. [Google Scholar] [CrossRef]
- De Loyola, M.B.; dos Reis, T.T.A.; de Oliveira, G.X.L.M.; da Fonseca, P.J.; Argañaraz, G.A.; Argañaraz, E.R. Alpha-1-antitrypsin: A possible host protective factor against Covid-19. Rev. Med Virol. 2020. [Google Scholar] [CrossRef]
- Sharif-Askari, N.S.; Sharif-Askari, F.S.; Alabed, M.; Temsah, M.H.; al Heialy, S.; Hamid, Q.; Halwani, R. Airways Expression of SARS-CoV-2 Receptor, ACE2, and TMPRSS2 Is Lower in Children Than Adults and Increases with Smoking and COPD. Mol. Ther. Methods Clin. Dev. 2020, 18. [Google Scholar] [CrossRef]
- Jose, R.J.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir. Med. 2020, 8, e46–e47. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Saftig, P.; Reiss, K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur. J. Cell Biol. 2011, 90. [Google Scholar] [CrossRef]
- Thabet, M.M.; Huizinga, T.W.J. Drug evaluation: Apratastat, a novel TACE/MMP inhibitor for rheumatoid arthritis. Curr. Opin. Investig. Drugs 2006, 7, 1014–1019. [Google Scholar]
- Friedman, S.; Levy, R.; Garrett, W.; Doval, D.; Bondarde, S.; Sahoo, T.; Lokanatha, D.; Julka, P.; Shenoy, K.; Nagarkar, R.; et al. Clinical Benefit of INCB7839, a Potent and Selective Inhibitor of ADAM10 and ADAM17, in Combination with Trastuzumab in Metastatic HER2 Positive Breast Cancer Patients. Poster Session Abstracts 2009, 69, 5056. [Google Scholar] [CrossRef]
- Maskos, K.; Fernandez-Catalan, C.; Huber, R.; Bourenkov, G.P.; Bartunik, H.; Ellestad, G.A.; Reddy, P.; Wolfson, M.F.; Rauch, C.T.; Castner, B.J.; et al. Crystal structure of the catalytic domain of human tumor necrosis factor-α-converting enzyme. Proc. Natl. Acad. Sci. USA 1998, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seegar, T.C.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638–1648.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caescu, C.I.; Jeschke, G.R.; Turk, B.E. Active-site determinants of substrate recognition by the metalloproteinases TACE and AD-AM10. Biochem. J. 2009, 424, 79–88. [Google Scholar] [CrossRef] [Green Version]
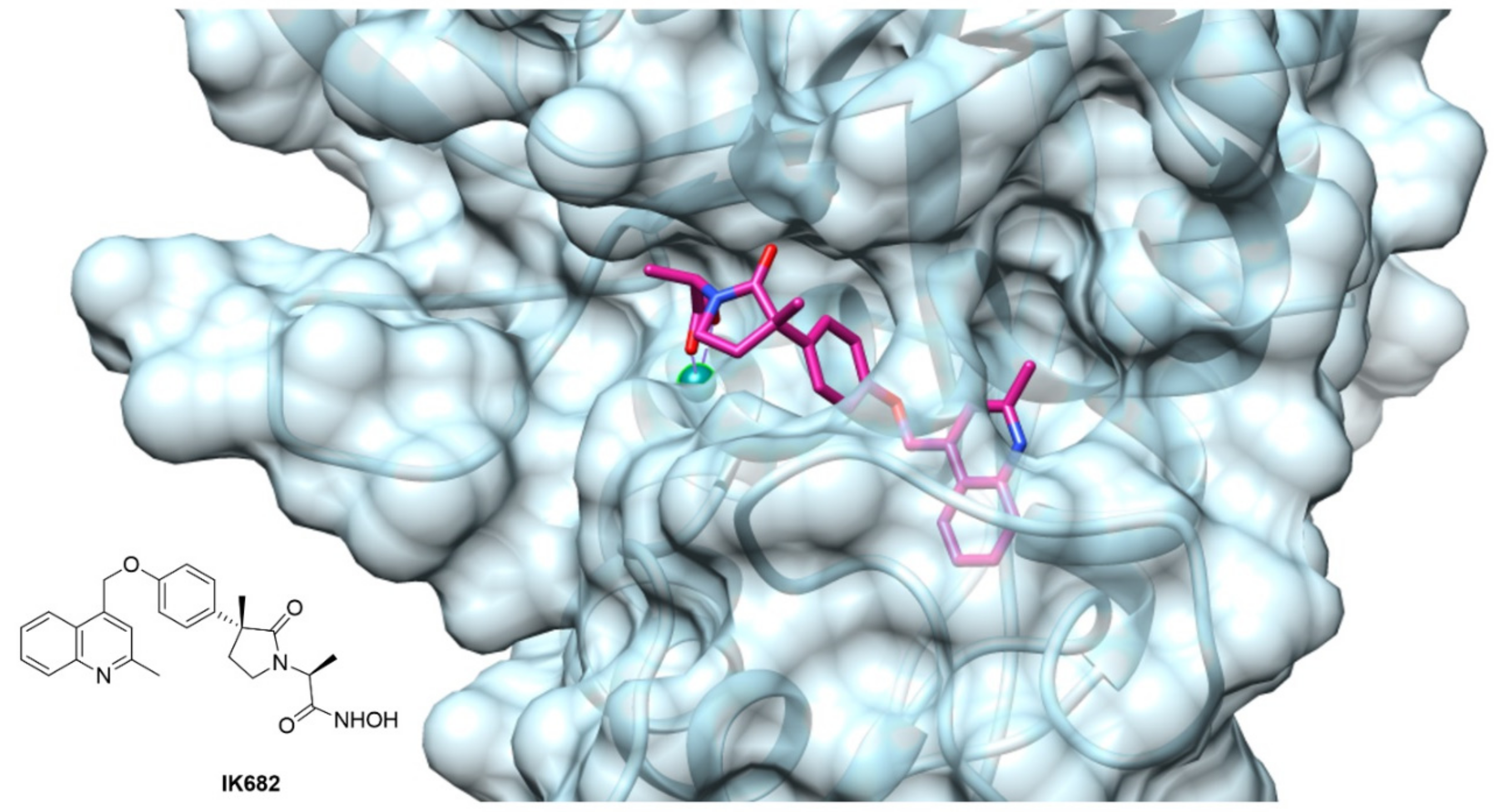
- Niu, X.; Umland, S.; Ingram, R.; Beyer, B.M.; Liu, Y.-H.; Sun, J.; Lundell, D.; Orth, P. IK682, a tight binding inhibitor of TACE. Arch. Biochem. Biophys. 2006, 451, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [Green Version]
- Murumkar, P.R.; Ghuge, R.B.; Chauhan, M.; Barot, R.R.; Sorathiya, S.; Choudhary, K.M.; Joshi, K.D.; Yadav, M.R. Recent developments and strategies for the discovery of TACE inhibitors. Expert Opin. Drug Discov. 2020, 15, 779–801. [Google Scholar] [CrossRef]
- Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A. ADAM Metalloproteinases as Potential Drug Targets. Curr. Med. Chem. 2019, 26, 2661–2689. [Google Scholar] [CrossRef]
- Ouvry, G.; Berton, Y.; Bhurruth-Alcor, Y.; Bonnary, L.; Bouix-Peter, C.; Bouquet, K.; Bourotte, M.; Chambon, S.; Comino, C.; Deprez, B.; et al. Identification of novel TACE inhibitors compatible with topical application. Bioorganic Med. Chem. Lett. 2017, 27, 1848–1853. [Google Scholar] [CrossRef]
- Boiteau, J.-G.; Ouvry, G.; Arlabosse, J.-M.; Astri, S.; Beillard, A.; Bhurruth-Alcor, Y.; Bonnary, L.; Bouix-Peter, C.; Bouquet, K.; Bourotte, M.; et al. Discovery and process development of a novel TACE inhibitor for the topical treatment of psoriasis. Bioorganic Med. Chem. 2018, 26, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Hirata, S.; Murata, T.; Suzuki, D.; Nakamura, S.; Jono-Ohnishi, R.; Hirose, H.; Sawaguchi, A.; Nishimura, S.; Sugimoto, N.; Eto, K. Selective Inhibition of ADAM17 Efficiently Me-diates Glycoprotein Ibα Retention During Ex Vivo Generation of Human Induced Pluripotent Stem Cell-Derived Platelets. STEM Cells Transl. Med. 2017, 6, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Laronha, H.; Carpinteiro, I.; Portugal, J.; Azul, A.; Polido, M.; Petrova, K.T.; Salema-Oom, M.; Caldeira, J. Challenges in Matrix Metalloproteinases Inhibition. Biomol. 2020, 10, 717. [Google Scholar] [CrossRef] [PubMed]
- Girijavallabhan, V.M.; Chen, L.; Dai, C.; Feltz, R.J.; Firmansjah, L.; Li, D.; Kim, S.H.; Kozlowski, J.A.; Lavey, B.J.; Kosinski, A.; et al. Novel TNF-α converting enzyme (TACE) inhibitors as potential treatment for inflammatory diseases. Bioorganic Med. Chem. Lett. 2010, 20, 7283–7287. [Google Scholar] [CrossRef]
- Tong, L.; Kim, S.H.; Rosner, K.; Yu, W.; Shankar, B.B.; Chen, L.; Li, D.; Dai, C.; Girijavallabhan, V.; Popovici-Muller, J.; et al. Fused bi-heteroaryl substituted hydantoin compounds as TACE inhibitors. Bioorganic Med. Chem. Lett. 2017, 27, 3037–3042. [Google Scholar] [CrossRef]
- Tong, L.; Kim, S.H.; Chen, L.; Kosinski, A.; Shankar, B.B.; Girijavallabhan, V.; Yang, D.-Y.; Yu, W.; Zhou, G.; Shih, N.-Y.; et al. Development of a prodrug of hydantoin based TACE inhibitor. Bioorganic Med. Chem. Lett. 2017, 27, 3704–3708. [Google Scholar] [CrossRef]
- Maekawa, M.; Tadaki, H.; Tomimoto, D.; Okuma, C.; Sano, R.; Ishii, Y.; Katsuda, Y.; Yoshiuchi, H.; Kakefuda, R.; Ohta, T.; et al. A Novel TNF-α Converting Enzyme (TACE) Selective Inhibitor JTP-96193 Prevents Insulin Resistance in KK-A y Type 2 Diabetic Mice and Diabetic Peripheral Neuropathy in Type 1 Diabetic. Mice. Biol. Pharm. Bull. 2019, 42, 1906–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.-H.; Liu, L.-J.; Lu, L.; He, B.; Kwong, D.W.J.; Wong, C.-Y.; Ma, D.-L. A metal-based tumour necrosis factor-alpha converting enzyme inhibitor. Chem. Commun. 2015, 51, 3973–3976. [Google Scholar] [CrossRef]
- Sarkate, A.; Murumkar, P.R.; Lokwani, D.; Kandhare, A.D.; Bodhankar, S.L.; Shinde, D.; Bothara, K. Design of selective TACE inhibitors using molecular docking studies: Synthesis and preliminary evaluation of anti-inflammatory and TACE inhibitory activity. SAR QSAR Environ. Res. 2015, 26, 905–923. [Google Scholar] [CrossRef]
- Minond, D.; Cudic, M.; Bionda, N.; Giulianotti, M.; Maida, L.; Houghten, R.A.; Fields, G.B. Discovery of Novel Inhibitors of a Disintegrin and Metalloprotease 17 (ADAM17) Using Glycosylated and Non-glycosylated Substrates. J. Biol. Chem. 2012, 287, 36473–36487. [Google Scholar] [CrossRef] [Green Version]
- Knapinska, A.M.; Dreymueller, D.; Ludwig, A.; Smith, L.; Golubkov, V.; Sohail, A.; Fridman, R.; Giulianotti, M.; LaVoi, T.M.; Houghten, R.A.; et al. SAR Studies of Exosite-Binding Substrate-Selective Inhibitors of A Disintegrin And Metalloprotease 17 (ADAM17) and Application as Selective in Vitro Probes. J. Med. Chem. 2015, 58, 5808–5824. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-D.; Zhao, C.-H.; Ding, H.-W.; Wu, Q.; Ren, T.-S.; Wang, J.; Chen, C.-Q.; Zhao, Q. A novel inhibitor of ADAM17 sensitizes colorectal cancer cells to 5-Fluorouracil by reversing Notch and epithelial-mesenchymal transition in vitro and in vivo. Cell Prolif. 2018, 51, e12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, D.; Jiang, Q.; Cao, S.; Sun, H.; Chai, Y.; Li, X.; Ren, T.; Yang, R.; Feng, F.; et al. Novel ADAM-17 inhibitor ZLDI-8 enhances the in vitro and in vivo chemotherapeutic effects of Sorafenib on hepatocellular carcinoma cells. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.Y.; Chu, H.X.; Tan, Y.X.; Qin, X.C.; Liu, M.Y.; Li, J.D.; Ren, T.S.; Zhang, Y.S.; Zhao, Q.C. Novel ADAM-17 inhibitor ZLDI-8 inhibits the metastasis of hepato-cellular carcinoma by reversing epithelial-mesenchymal transition in vitro and in vivo. Life Sci. 2020, 244. [Google Scholar] [CrossRef]
- Lu, H.Y.; Zu, Y.X.; Jiang, X.W.; Sun, X.T.; Liu, T.Y.; Li, R.L.; Wu, Q.; Zhang, Y.S.; Zhao, Q.C. Novel ADAM-17 inhibitor ZLDI-8 inhibits the proliferation and metastasis of chemo-resistant non-small-cell lung cancer by reversing Notch and epithelial mesenchymal transition in vitro and in vivo. Pharmacol. Res. 2019, 148. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, P.E.; Solomon, A.; Miller, A.B.; Leesnitzer, M.A.; Sagi, I.; Milla, M.E. Inhibition of the Tumor Necrosis Factor-α-converting Enzyme by Its Pro Domain. J. Biol. Chem. 2004, 279, 31638–31645. [Google Scholar] [CrossRef] [Green Version]
- Geribaldi-Doldán, N.; Carrasco, M.; Murillo-Carretero, M.; Domínguez-García, S.; García-Cózar, F.J.; Muñoz-Miranda, J.P.; el Río-García, V.; Verástegui, C.; Castro, C. Specific inhibition of ADAM17/TACE promotes neurogenesis in the injured motor cortex. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Wong, E.; Cohen, T.; Romi, E.; Levin, M.; Peleg, Y.; Arad, U.; Yaron, A.; Milla, M.E.; Sagi, I. Harnessing the natural inhibitory domain to control TNFα Con-verting Enzyme (TACE) activity In Vivo. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Gamez, A.; Chen, D.; Nabuurs, A.G.; Quax, W.J.; DeMaria, M.; Boersma, Y.L. A Bispecific Inhibitor of the EGFR/ADAM17 Axis Decreases Cell Proliferation and Migration of EGFR-Dependent Cancer Cells. Cancers 2020, 12, 411. [Google Scholar] [CrossRef] [Green Version]
- Solomon, K.A.; Pesti, N.; Wu, G.; Newton, R.C. Cutting edge: A dominant negative form of TNF-alpha converting enzyme inhibits proTNF and TNFRII secretion. J. Immunol. 1999, 163, 4105–4108. [Google Scholar] [PubMed]
- Itoh, Y.; Ito, N.; Nagase, H.; Seiki, M. The Second Dimer Interface of MT1-MMP, the Transmembrane Domain, Is Essential for ProMMP-2 Activation on the Cell Surface. J. Biol. Chem. 2008, 283, 13053–13062. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE Activation by MAPK-Mediated Regulation of Cell Surface Dimerization and TIMP3 Association. Sci. Signal. 2012, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Maskos, K.; Knauper, V.; Dodds, P.; Murphy, G. Mapping and characterization of the functional epitopes of tissue in-hibitor of metalloproteinases (TIMP)-3 using TIMP-1 as the scaffold: A new frontier in TIMP engineering. Protein Sci. 2002, 11, 2493–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Verma, V.; Maskos, K.; Nath, D.; Knäuper, V.; Dodds, P.; Amour, A.; Murphy, G. Engineering N-terminal domain of tissue inhibitor of met-alloproteinase (TIMP)-3 to be a better inhibitor against tumour necrosis factor-α-converting enzyme. Biochem. J. 2002, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Kashiwagi, M.; Kota, S.; Xie, Z.; Nagase, H.; Brew, K. Reactive site mutations in tissue inhibitor of metalloproteinase-3 dis-rupt inhibition of matrix metalloproteinases but not tumor necrosis factor-α-converting enzyme. J. Biol. Chem. 2005, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, N.H.; Kashiwagi, M.; Visse, R.; Jones, J.; Enghild, J.J.; Brew, K.; Nagase, H. Reactive-site mutants of N-TIMP-3 that selectively inhibit ADAMTS-4 and ADAMTS-5: Biological and structural implications. Biochem. J. 2010, 431, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Vo, P.; Kanakis, I.; Liu, K.; Bou-Gharios, G. Aggrecanase-selective tissue inhibitor of metalloproteinase-3 (TIMP3) protects articular cartilage in a surgical mouse model of osteoarthritis. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Tape, C.J.; Willems, S.H.; Dombernowsky, S.L.; Stanley, P.L.; Fogarasi, M.; Ouwehand, W.; McCafferty, J.; Murphy, G. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA 2011, 108, 5578–5583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Cook, K.; Xu, L.; Cheng, L.; Damschroder, M.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Molecular basis for the mechanism of action of an anti-TACE antibody. mAbs 2016, 8, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Rios-Doria, J.; Sabol, D.; Chesebrough, J.; Stewart, D.; Xu, L.; Tammali, R.; Cheng, L.; Du, Q.; Schifferli, K.; Rothstein, R.; et al. Amonoclonal antibody to ADAM17 inhibits tumor growth by inhibiting EGFR and non-EGFR-mediated pathways. Mol. Cancer Ther. 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, X.; Chinenov, Y.; Redecha, P.; Madaio, M.; Roelofs, J.J.; Farber, G.; Issuree, P.D.; Donlin, L.; Mcllwain, D.R.; Mak, T.W.; et al. iRhom2 promotes lupus nephritis through TNF-α and EGFR signaling. J. Clin. Investig. 2018, 128, 1397–1412. [Google Scholar] [CrossRef] [PubMed]
- Haxaire, C.; Hakobyan, N.; Pannellini, T.; Carballo, C.; McIlwain, D.; Mak, T.W.; Rodeo, S.; Acharya, S.; Li, D.; Szymonifka, J.; et al. Blood-induced bone loss in murine hemo-philic arthropathy is prevented by blocking the iRhom2/ADAM17/TNF-a pathway. Blood 2018, 132. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Cell-to-Cell Communication | Signaling- Receptors | Cell Adhesion | Cellular Transport | Enzyme | Others |
|---|---|---|---|---|---|---|
| TNFα | Amphiregulin | Axl | ALCAM | SCRB1 | ACE-2 | APP |
| CSF-1 | HB-EGF | CD16 | CD44 | LRP-1 | Carbonic Hydrolase 9 | APLP-2 |
| KL-1 | TGFα | CD163 | L-selectin | LDL-R | Klotho | Prion protein |
| KL-2 | Epigen | CD30 (TNFRSF8) | Collagen XVII | SORCS-1 | NPR1 | Vasorin |
| Lymphotoxin α | Epiregulin | CD40 (TNFRSF5) | Desmoglein-2 | SORCS-3 | PMEL-17 | |
| RANKL | NRG-1 | CD89 | EpCam | SORL-1 | Sydecan-1 | |
| Cx3cl1 | Jagged | EPCR | GP-1ba | SORT-1 | Sydecan-4 | |
| IL-8 | DLL-1 | ErbB-4 | GP-5 | TREM-2 | Pre-adypocyte factor | |
| PD-L1 | GHRH receptor | GP-6 | IGF-2R | Collagen IV * | ||
| ICOS-L | M-CSFR | ICAM-1 | PCPE-1 * | |||
| IL-15R | Notch-1 | JAM-A | cystatin C * | |||
| IL-1R2 | NRP-1 | L1-CAM | Ebola virus Glycoprotein ** | |||
| IL-6R | PTK7 | LYPD3 | ||||
| LAG-3 | PTPRZ | MUC-1 | ||||
| MIC-A | PTPRF | NCAM | ||||
| MIC-B | SEMA-4D | Nectin-4 | ||||
| TIM-1 | TNF-R1 | SynCAM-1 | ||||
| TIM-3 | TNF-R2 | VACM-1 | ||||
| TIM-4 | NTRK1 | Thrombospondin-4 | ||||
| VEGF-R2 |
| Pathology | Role | Effects of ADAM17 Inactivation | Substrates | Reference |
|---|---|---|---|---|
| Rheumatoid Arthritis | Detrimental in the progression of the disease | Decreased inflammation and cartilage breakdown | TNF IL-6R | [95,103] |
| Regulates the release of TNF and activation of TNFR signaling | ||||
| Regulates the activation of the pro-inflammatory IL-6 trans-signaling | ||||
| Osteoarthritis | Predicted negative role in the progression of osteoarthritis | Enhanced secretion of metalloproteases and lowered deposition of ECM | TNF LRP-1 | [108,109,117] |
| Regulates TNF signaling | ||||
| Controls levels of the endocytic receptor LRP-1 | Decreased metalloprotease turnover | |||
| Lung pathology | Negative role in the disease | Lower neutrophil recruitment and inflammation | TNF L-selectin CX3CL1 JAM-A | [119,120] |
| ADAM17 in leukocytes controls levels of alveolar TNF and L-selectin | ||||
| ADAM17 in endothelial cells regulates adhesion molecules and chemokines | ||||
| Nerve recovery | Negative role in nerve regeneration | Higher clearance of apoptotic cells and augmented phagocytic capability of microglia | TREM2 | [148,150] |
| ADAM17 in microglia controls TREM2 levels and phagocytosis | ||||
| Cancer | ADAM17 is associated with cancer progression | Reduced cancer progression | TGFα HB-EGF | [153,156,159, 160] |
| Regulates EGFR signaling and proliferation of cancer cells | Reduced T cell activation and their ability to kill tumors | |||
| PD-L1 CD16A | ||||
| Modulates the cleavage of PD-L1 and immuno evasion of cancer cells | ||||
| COVID-19 | Unclear whether ADAM17 activity is beneficial or detrimental | ACE2 TNF | [166,167,168] | |
| Regulates levels of ACE2 and SARS-CoV-2 entry | In vivo models currently not available | |||
| Promotes TNF release and the “cytokine storm” | ||||
| Alzheimer′s Disease | ADAM17 has opposite effects in the development of AD depending on its tissue expression | Decrease of neuroinflammation and phagocytic capability of microglia | TNF TREM2 | [135,136,144] |
| ADAM17 in microglia regulates TNFR signaling and TREM2 levels | Activation of the anti-amyloidgenic processing of APP and reduced plaque deposition | |||
| APP | ||||
| ADAM17 in neurons regulates shedding of APP | ||||
| Inflammatory bowel disease | Protective role in the development of ulcerative colitis | Diminished EGFR activation, epithelial cell growth and goblet cell differentiation | TGFα | [16,128] |
| Controls EGFR signaling | ||||
| Atherosclerosis | Protective role in the progression of atherosclerosis | Enhanced TNFR2 signaling, proliferation of macrophages and their augmented recruitment to the lesion sites | TNF TNFR2 | [123] |
| Regulates TNFR signaling |
| Code | Structure | ADAM17 | ADAM10 | Ref |
|---|---|---|---|---|
| 1 |  | 12 | >10000 | [183] |
| 2 |  | 4 | 950 | [184] |
| 3 (KP475) |  | 11 | 748 | [185] |
| 4 |  | 0.62 (Ki) | 22 | [186] |
| 5 |  | 0.5 (Ki) | -a | [187] |
| 6 |  | - | - | [188] |
| 7 (JTP-96193) |  | 5.4 | >10000 | [189] |
| 8 |  | 28000 | - | [190] |
| 9 |  | - | - | [191] |
| 10 |  | 4200 | >100000 | [192,193] |
| 11 (ZLDI-8) |  | 72000 | - | [194,195] |
| Compound | Effect | Cell Line | IC50 | In Vivo/Ex Vivo Assay |
|---|---|---|---|---|
| 1 | Inhibition of TNFα release | Human Keratinocytes (NHEK) | 1.3 µM | - |
| 2 | Inhibition of TNFα release | NHEK | 3 nM | Strong activity in a mouse model of oxazolone- induced chronic skin inflammation |
| 3 (KP475) | Inhibition of GPIbα shedding | human iPSC platelets | ≈ 100 nM | Positive results in a thrombus formation model using immunodeficient mice |
| 5 | Inhibition of TNFα production | human whole blood (hWBA): | 287 nM | - |
| 6 | Inhibition of TNFα production | human whole blood (hWBA): | 281 nM | Good PK properties in rats and monkeys |
| 7 (JTP-96193) | Inhibition of TNFα release | rat whole blood (rWBA) | 170 nM | In mouse models of obesity, it reduced the TNF-α release from the fat tissue and prevented development of diabetes; in mouse models of diabetes, it improved insulin resistance |
| 8 | Inhibition of TNFα release | PMA-differentiated THP-1 cells | 11.24 µM | - |
| 9 | Inhibition of TNFα release | Rat serum | 68.48% inhibition at 20 mg/kg dose | Good anti- inflammatory activity in carrageenan-induced rat paw edema model |
| 10 | Inhibition of TNF-α cleavage | THP-1 cells | 100 µM | - |
| Inhibition of Heregulin cleavage | ||||
| A549 cells | 100% inhibition at 40 µM | |||
| 11 (ZLDI-8) | Inhibition of Notch signaling pathway | LoVo cells | 5.57 μM | 11 enhanced the effect of Sorafenib on inhibiting tumor growth in a nude HCC-bearing mice model [194] |
| SW480 cells | 7.42 μM | |||
| MHCC97-H cells | 5.32 μM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calligaris, M.; Cuffaro, D.; Bonelli, S.; Spanò, D.P.; Rossello, A.; Nuti, E.; Scilabra, S.D. Strategies to Target ADAM17 in Disease: From Its Discovery to the iRhom Revolution. Molecules 2021, 26, 944. https://doi.org/10.3390/molecules26040944
Calligaris M, Cuffaro D, Bonelli S, Spanò DP, Rossello A, Nuti E, Scilabra SD. Strategies to Target ADAM17 in Disease: From Its Discovery to the iRhom Revolution. Molecules. 2021; 26(4):944. https://doi.org/10.3390/molecules26040944
Chicago/Turabian StyleCalligaris, Matteo, Doretta Cuffaro, Simone Bonelli, Donatella Pia Spanò, Armando Rossello, Elisa Nuti, and Simone Dario Scilabra. 2021. "Strategies to Target ADAM17 in Disease: From Its Discovery to the iRhom Revolution" Molecules 26, no. 4: 944. https://doi.org/10.3390/molecules26040944
APA StyleCalligaris, M., Cuffaro, D., Bonelli, S., Spanò, D. P., Rossello, A., Nuti, E., & Scilabra, S. D. (2021). Strategies to Target ADAM17 in Disease: From Its Discovery to the iRhom Revolution. Molecules, 26(4), 944. https://doi.org/10.3390/molecules26040944