Four Isotope-Labeled Recombination Pathways of Ozone Formation


Abstract
:1. Introduction
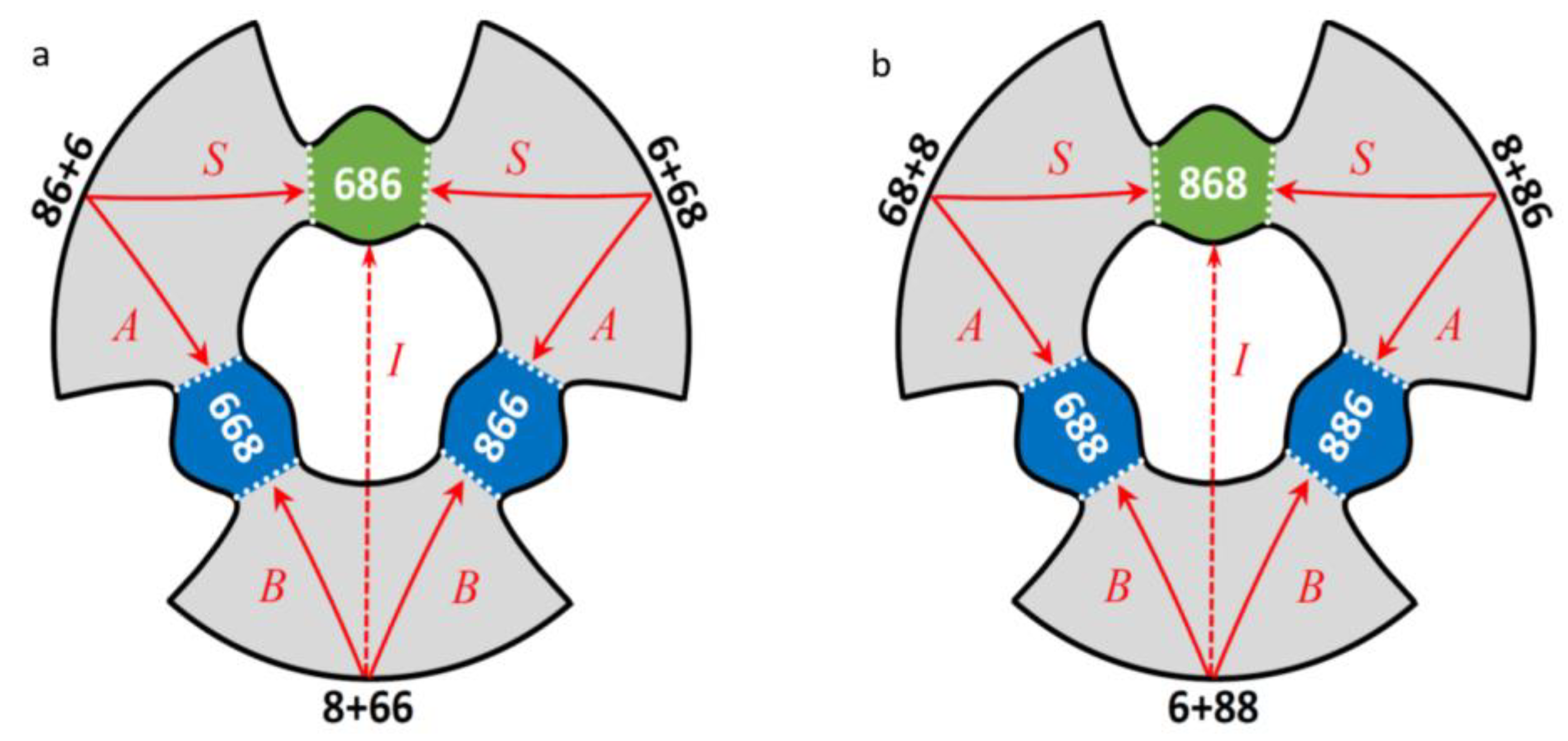
| A: | 16O + 16O18O 16O16O18O | (1) |
| B: | 18O + 16O16O 18O16O16O | (2) |
| S: | 16O + 18O16O 16O18O16O | (3) |
| I: | 18O + 16O16O 16O18O16O | (4) |
| A: | 18O + 18O16O 18O18O16O | (5) |
| B: | 16O + 18O18O 16O18O18O | (6) |
| S: | 18O + 16O18O 18O16O18O | (7) |
| I: | 16O + 18O18O 18O16O18O | (8) |
- (1)
- Quantum ΔZPE-effect, responsible for large difference, about 60%, between 2κΑ and κB of the pathways A and B that are just slightly endo/exothermic (one with respect to another). This phenomenon will be named ζ-effect.
- (2)
- (3)
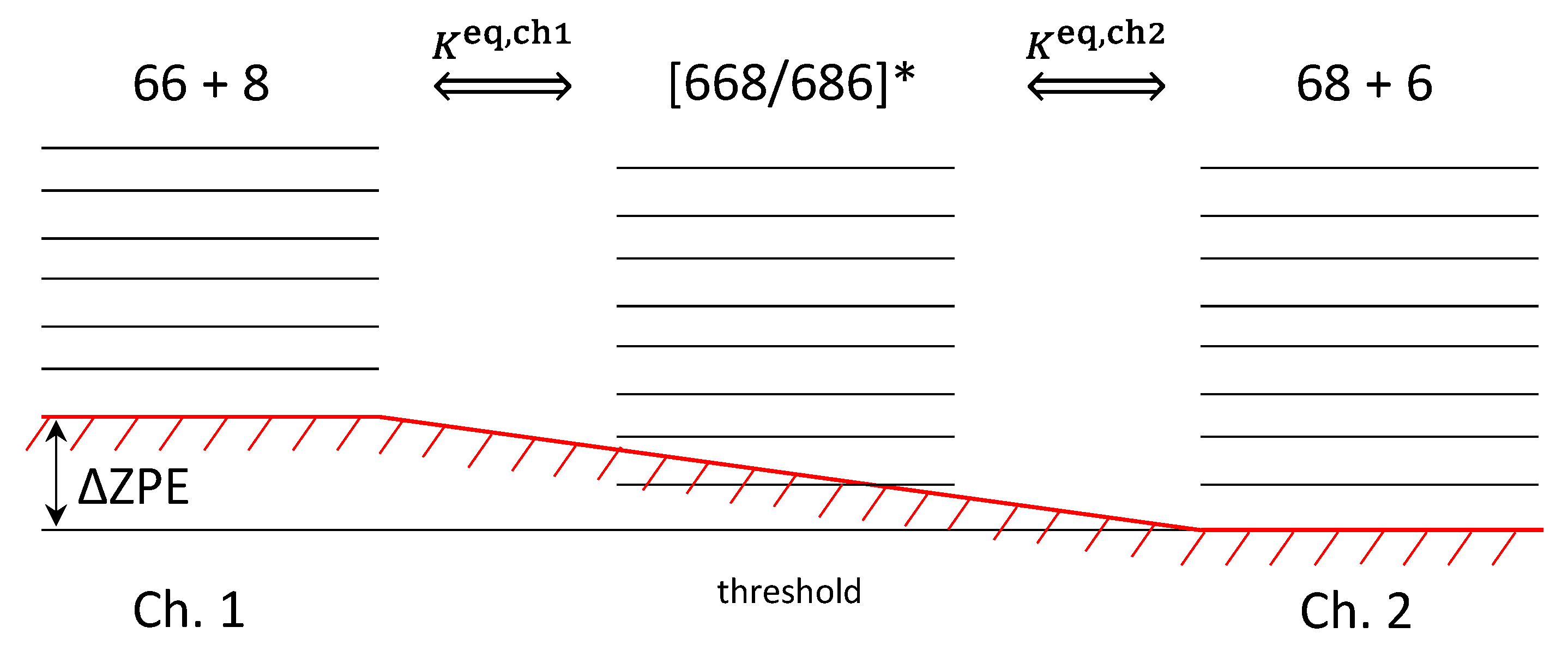
- Very large difference, by a factor of ~5, between the insertion rate coefficients κI in the singly and doubly substituted cases [1]. Here we will call it ξ-effect.
2. Theory
2.1. The Four Pathways of the Recombination Reaction

2.2. Kinetic Weight of a Resonance and the Dynamical Partition Function
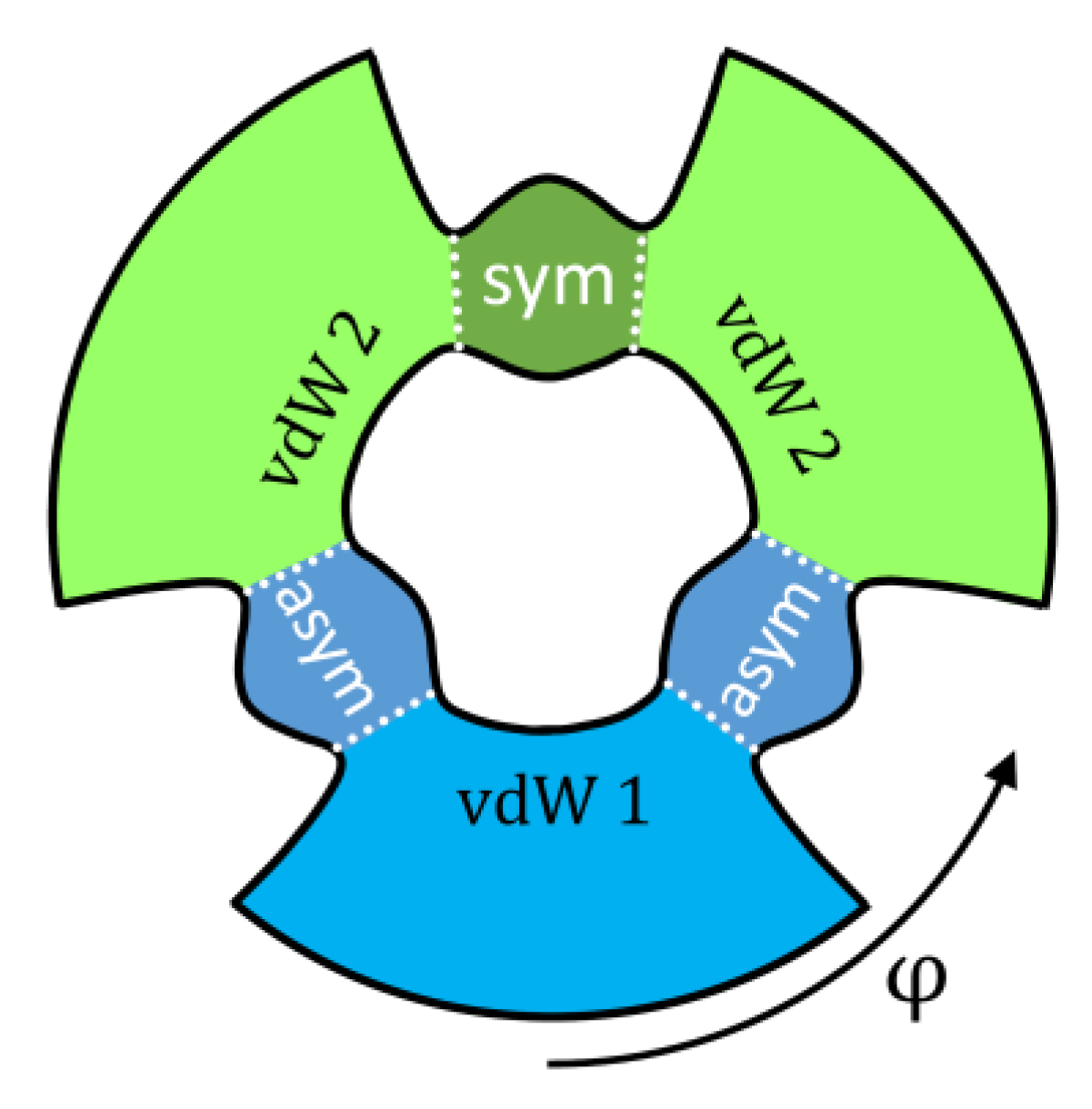
2.3. Formation of Symmetric and Asymmetric Molecules
3. Results and Discussion
3.1. Pathway-Specific Rate Coefficients
3.2. Isotope Effects
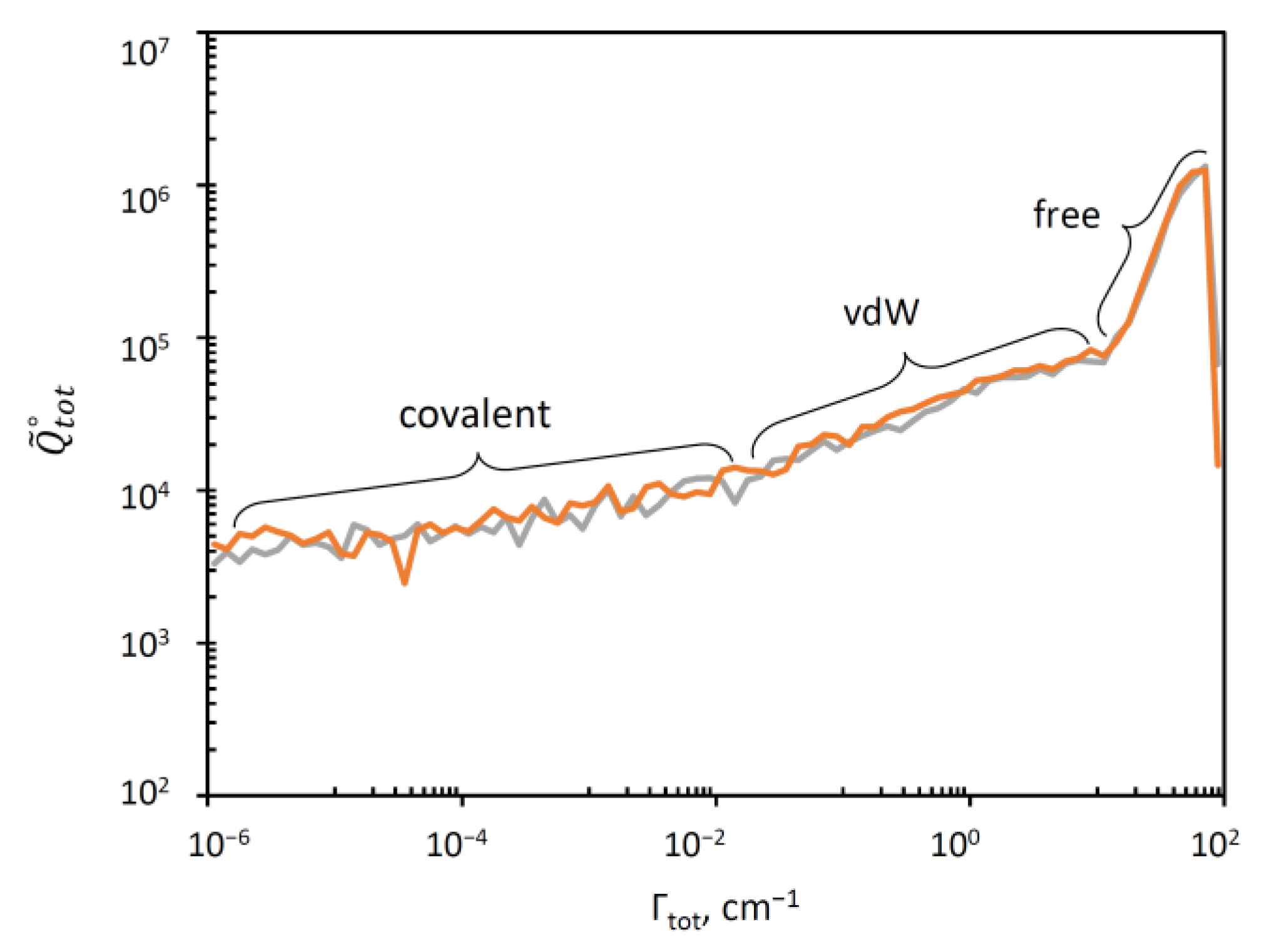
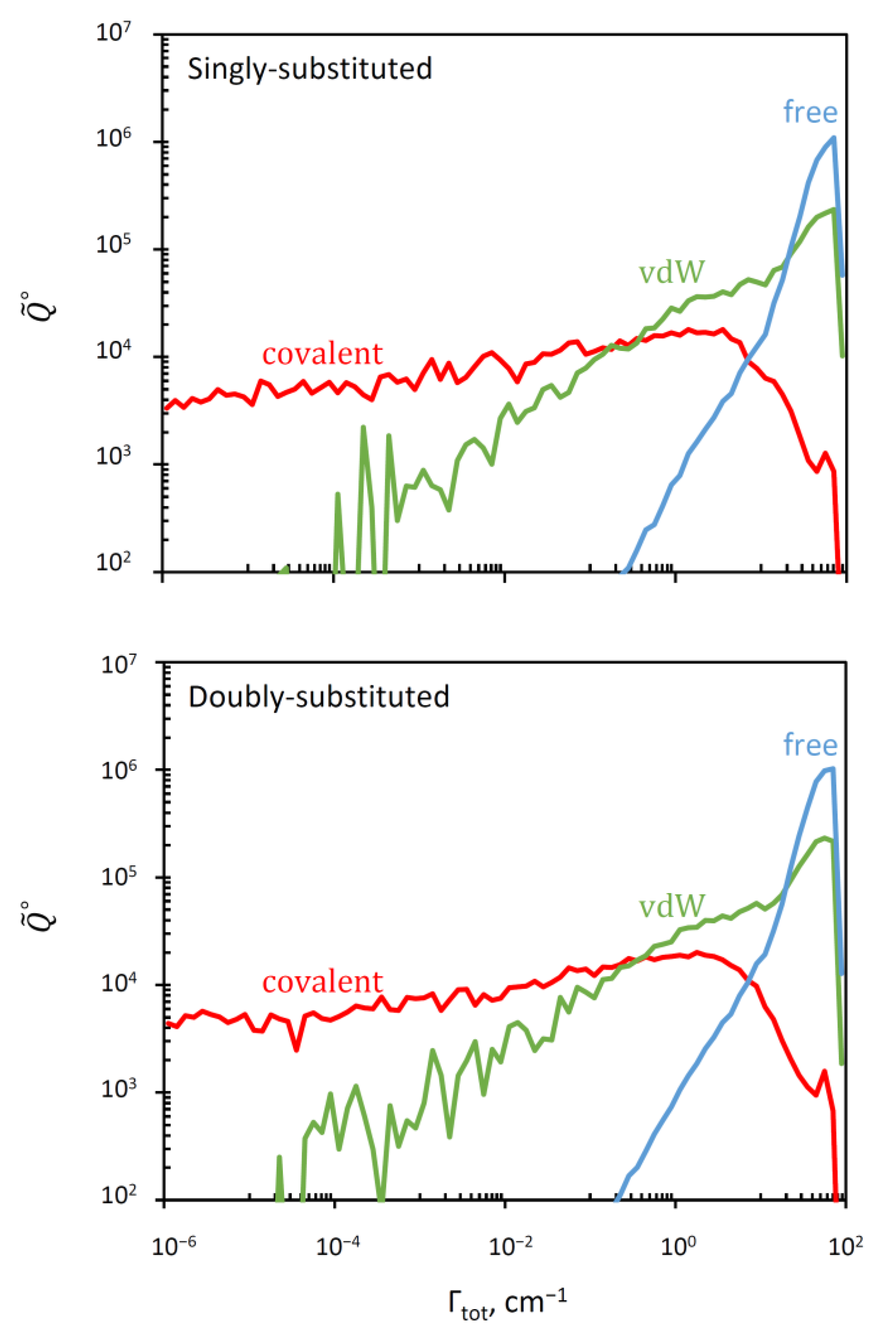
3.3. Distribution of Resonance Widths
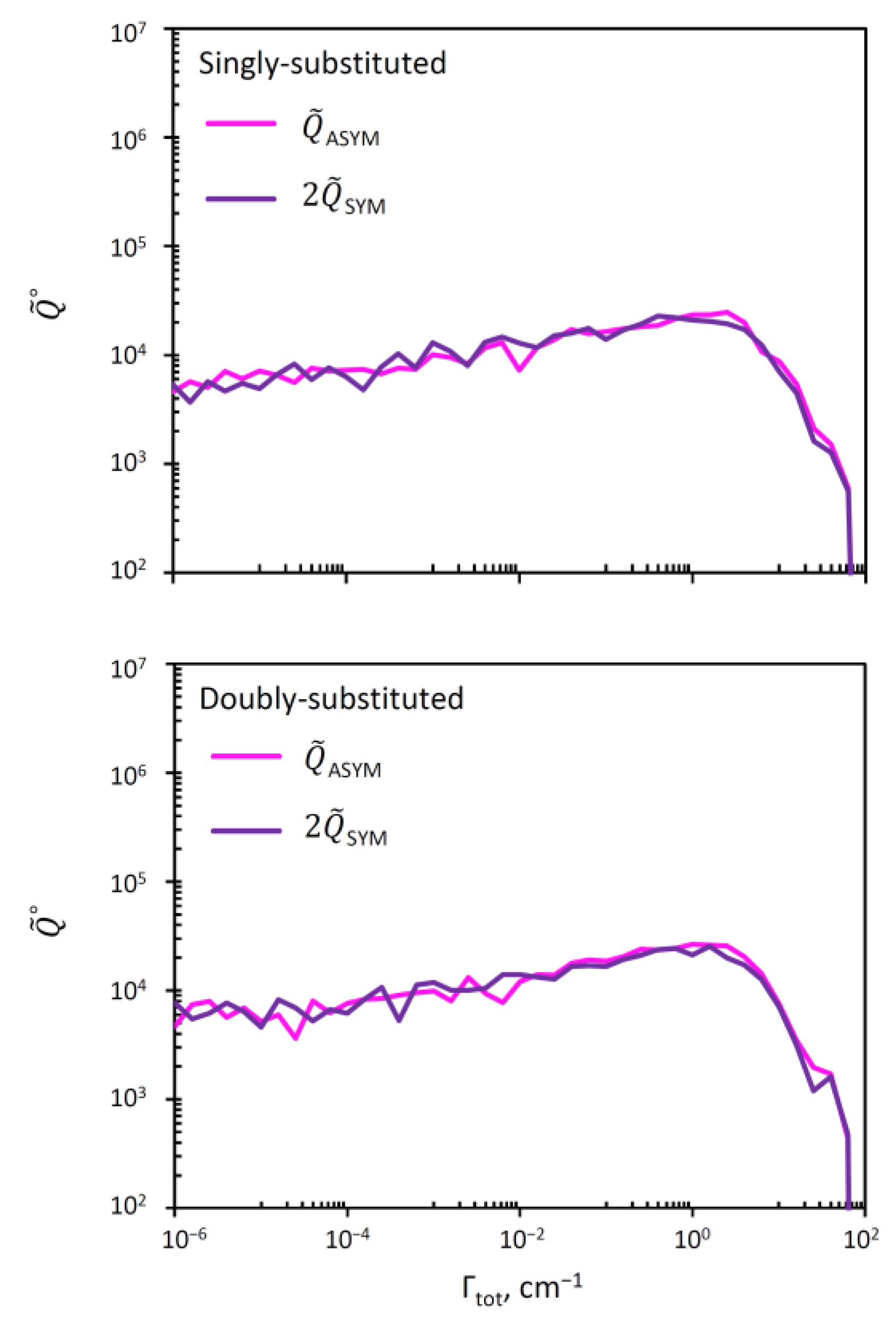
3.4. Resonance Widths in Symmetric and Asymmetric Ozone Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Janssen, C.; Guenther, J.; Mauersberger, K.; Krankowsky, D. Kinetic origin of the ozone isotope effect: A critical analysis of enrichments and rate coefficients. Phys. Chem. Chem. Phys. 2001, 3, 4718–4721. [Google Scholar] [CrossRef]
- Mauersberger, K.; Erbacher, B.; Krankowsky, D.; Günther, J.; Nickel, R. Ozone isotope enrichment: Isotopomer-specific rate coefficients. Science 1999, 283, 370–372. [Google Scholar] [CrossRef]
- Janssen, C.; Guenther, R.; Krankowsky, D.; Mauersberger, K. Temperature dependence of ozone rate coefficients and isotopologue fractionation in 16O–18O oxygen mixtures. Chem. Phys. Lett. 2003, 367, 34–38. [Google Scholar] [CrossRef]
- Guenther, J.; Erbacher, B.; Krankowsky, D.; Mauersberger, K. Pressure dependence of two relative ozone formation rate coefficients. Chem. Phys. Lett. 1999, 306, 209–221. [Google Scholar] [CrossRef]
- Mauersberger, K.; Krankowsky, D.; Janssen, C. Oxygen isotope processes and transfer reactions. Space Sci. Rev. 2003, 106, 265–279. [Google Scholar] [CrossRef]
- Janssen, C. Intramolecular isotope distribution in heavy ozone (16O18O16O and 16O16O18O). J. Geophys. Res. 2005, 110, D08308. [Google Scholar]
- Janssen, C. Investigation and assessment of an oxygen isotope anomaly. In Habilitation; University of Heidelberg: Heidelberg, Germany, 2004. [Google Scholar]
- Tuzson, B. Symmetry Specific Study of Ozone Isotopomer Formation. Ph. D. Thesis, University of Heidelberg, Heidelberg, Germany, 2005. [Google Scholar]
- Guenther, J.; Krankowsky, D.; Mauersberger, K. Third-body dependence of rate coefficients for ozone formation in 16O–18O mixtures. Chem. Phys. Lett. 2000, 324, 31–36. [Google Scholar] [CrossRef]
- Hathorn, B.C.; Marcus, R.A. An intramolecular theory of the mass independent isotope effect for ozone. I. J. Chem. Phys. 1999, 111, 4087–4100. [Google Scholar] [CrossRef]
- Hathorn, B.C.; Marcus, R.A. An Intramolecular theory of the mass-independent isotope effect for ozone. II. Numerical implementation at low pressures using a loose transition state. J. Chem. Phys. 2000, 113, 9497–9509. [Google Scholar] [CrossRef] [Green Version]
- Janssen, C.; Marcus, R.A. Does symmetry drive isotopic anomalies in ozone isotopomer formation? Science 2001, 294, 951a. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.Q.; Marcus, R.A. Strange and unconventional isotope effects in ozone formation. Science 2001, 293, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babikov, D.; Kendrick, B.K.; Walker, R.B.; Pack, R.T.; Fleurat-Lesard, P.; Schinke, R. Formation of ozone: Metastable states and anomalous isotope effect. J. Chem. Phys. 2003, 119, 2577–2589. [Google Scholar] [CrossRef]
- Babikov, D.; Kendrick, B.K.; Walker, R.B.; Pack, R.T.; Fleurat-Lesard, P.; Schinke, R. Metastable states of ozone calculated on an accurate potential energy surface. J. Chem. Phys. 2003, 118, 6298–6308. [Google Scholar] [CrossRef]
- Babikov, D.; Kendrick, B.K.; Walker, R.B.; Schinke, R.; Pack, R.T. Quantum origin of an anomalous isotope effect in ozone formation. Chem. Phys. Lett. 2003, 372, 686–691. [Google Scholar] [CrossRef]
- Grebenshchikov, S.Y.; Schinke, R. Towards quantum mechanical description of the unconventional mass-dependent isotope effect in ozone: Resonance recombination in the strong collision approximation. J. Chem. Phys. 2009, 131, 181103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryvohuz, M.; Marcus, R.A. Coriolis coupling as a source of non-RRKM effects in triatomic near-symmetric top molecules: Diffusive intramolecular energy exchange between rotational and vibrational degrees of freedom. J. Chem. Phys. 2010, 132, 224304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryvohuz, M.; Marcus, R.A. Coriolis coupling as a source of non-RRKM effects in ozone molecule: Lifetime statistics of vibrationally excited ozone molecules. J. Chem. Phys. 2010, 132, 224305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, M.V.; Babikov, D. On molecular origin of mass-independent fractionation of oxygen isotopes in the ozone forming recombination reaction. Proc. Nat. Acad. Sci. USA 2013, 110, 17708–17713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teplukhin, A.; Babikov, D. Several levels of theory for description of isotope effects in ozone: Symmetry effect and mass effect. J. Phys. Chem. A 2018, 122, 9177–9190. [Google Scholar] [CrossRef] [Green Version]
- Teplukhin, A.; Gayday, I.; Babikov, D. Several levels of theory for description of isotope effects in ozone: Effect of resonance lifetimes and channel couplings. J. Chem. Phys. 2018, 149, 164302. [Google Scholar] [CrossRef] [Green Version]
- Gayday, I.; Teplukhin, A.; Kendrick, B.K.; Babikov, D. Theoretical treatment of the Coriolis effect using hyperspherical coordinates, with application to the ro-vibrational spectrum of ozone. J. Phys. Chem. A 2020, 124, 2808–2819. [Google Scholar] [CrossRef] [Green Version]
- Gayday, I.; Teplukhin, A.; Kendrick, B.K.; Babikov, D. The role of rotation–vibration coupling in symmetric and asymmetric isotopomers of ozone. J. Chem. Phys. 2020, 152, 144104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.M.; Houston, P.L. Theories and simulations of roaming. Chem. Soc. Rev. 2017, 46, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.H.; Lapierre, D.; Gatti, F.; Kokoouline, V.; Tyuterev, V.G. The role of ozone vibrational resonances in the isotope exchange reaction 16O16O + 18O → 18O16O + 16O: The time-dependent picture. J. Phys. Chem. A 2019, 123, 7733–7743. [Google Scholar] [CrossRef]
- Mauguière F. A., L.; Collins, P.; Stamatiadis, S.; Li, A.; Ezra G., S.; Farantos S., C.; Kramer Z., C.; Carpenter B., K.; Wiggins, S.; Guo, H. Toward understanding the roaming mechanism in H + MgH → Mg + HH reaction. J. Phys. Chem. A 2016, 120, 5145–5154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletti, C.; Billing, G. Vibrational energy transfer in molecular oxygen collisions. Chem. Phys. Lett. 2002, 356, 14–22. [Google Scholar] [CrossRef]
- Charlo, D.; Clary, D.C. Quantum-mechanical calculations on termolecular association reactions XY + Z + M→XYZ + M: Application to ozone formation. J. Chem. Phys. 2002, 117, 1660–1672. [Google Scholar] [CrossRef]
- Charlo, D.; Clary, D.C. Quantum-mechanical calculations on pressure and temperature dependence of three-body recombination reactions: Application to ozone formation rates. J. Chem. Phys. 2004, 120, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Bowman, J.M. Quantum inelastic scattering study of isotope effects in ozone stabilization dynamics. Chem. Phys. Lett. 2005, 412, 131–134. [Google Scholar] [CrossRef]
- Ivanov, M.V.; Babikov, D. On stabilization of scattering resonances in recombination reaction that forms ozone. J. Chem. Phys. 2016, 144, 154301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luther, K.; Oum, K.; Troe, J. The role of the radical-complex mechanism in the ozone recombination/dissociation reaction. Phys. Chem. Chem. Phys. 2005, 7, 2764–2770. [Google Scholar] [CrossRef] [PubMed]
- Bare, T.; Hase, W. Unimolecular reaction dynamics. In Theory and Experiment; Oxford University Press: New York, NY, USA, 1996. [Google Scholar]
- Ivanov, M.; Babikov, D. Collisional stabilization of van der Waals states in ozone. J. Chem. Phys. 2011, 134, 174308. [Google Scholar] [CrossRef] [PubMed]
- Gayday, I.; Grushnikova, E.; Babikov, D. Influence of the Coriolis effect on the properties of scattering resonances in symmetric and asymmetric isotopomers of ozone. Phys. Chem. Chem. Phys. 2020, 22, 27560–27571. [Google Scholar] [CrossRef] [PubMed]
- Kokoouline, V.; Lapierre, D.; Alijah, A.; Tyuterev, V. Localized and delocalized bound states of the main isotopologue 48O3 and of 18O-enriched 50O3 isotopomers of the ozone molecule near the dissociation threshold. Phys. Chem. Chem. Phys. 2020, 22, 15885–15899. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Robert, F. On the mass independent isotope fractionation in ozone. Chem. Phys. 2018, 513, 287–294. [Google Scholar] [CrossRef]
- Robert, F.; Baraut-Guinet, L.; Cartigny, P.; Reinhardt, P. An experimental test for the mass independent isotopic fractionation mechanism proposed for ozone. Chem. Phys. 2019, 523, 191–197. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rate Coefficients | Experiment [1,2,3,4,5,6,7,8] | Theory, This Work | ||
|---|---|---|---|---|
| Single | Double | Single | Double | |
| S | 60 | 62 | 60 | 61 |
| A | 86 | 55 | 73 | 57 |
| B | 55 | 90 | 55 | 73 |
| I | 0.36 | 1.74 | 0.18 | 1.39 |
| Isotope Effects | ||||
| ζ | 1.68 | 1.77 | 1.45 | 1.38 |
| η | 1.13 | 1.19 | 1.02 | 1.08 |
| 4.8 | 7.9 | |||
| Resonance Type | ||||
|---|---|---|---|---|
| Single | Double | Single | Double | |
| Covalent well | 1.00 | 1.01 | 33.4 | 33.0 |
| Van der Waals plateau | 27.9 | 26.9 | 1.20 | 1.24 |
| Asymptotic states (free) | 68.9 | 67.3 | 0.484 | 0.496 |
| Isotopomer of Ozone | ||||
|---|---|---|---|---|
| Single | Double | Single | Double | |
| Symmetric | 1.22 | 0.91 | 27.3 | 36.7 |
| Asymmetric | 1.17 | 0.94 | 28.5 | 35.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babikov, D.; Grushnikova, E.; Gayday, I.; Teplukhin, A. Four Isotope-Labeled Recombination Pathways of Ozone Formation. Molecules 2021, 26, 1289. https://doi.org/10.3390/molecules26051289
Babikov D, Grushnikova E, Gayday I, Teplukhin A. Four Isotope-Labeled Recombination Pathways of Ozone Formation. Molecules. 2021; 26(5):1289. https://doi.org/10.3390/molecules26051289
Chicago/Turabian StyleBabikov, Dmitri, Elizaveta Grushnikova, Igor Gayday, and Alexander Teplukhin. 2021. "Four Isotope-Labeled Recombination Pathways of Ozone Formation" Molecules 26, no. 5: 1289. https://doi.org/10.3390/molecules26051289
APA StyleBabikov, D., Grushnikova, E., Gayday, I., & Teplukhin, A. (2021). Four Isotope-Labeled Recombination Pathways of Ozone Formation. Molecules, 26(5), 1289. https://doi.org/10.3390/molecules26051289