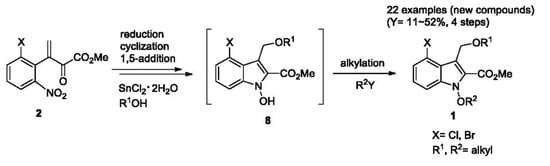
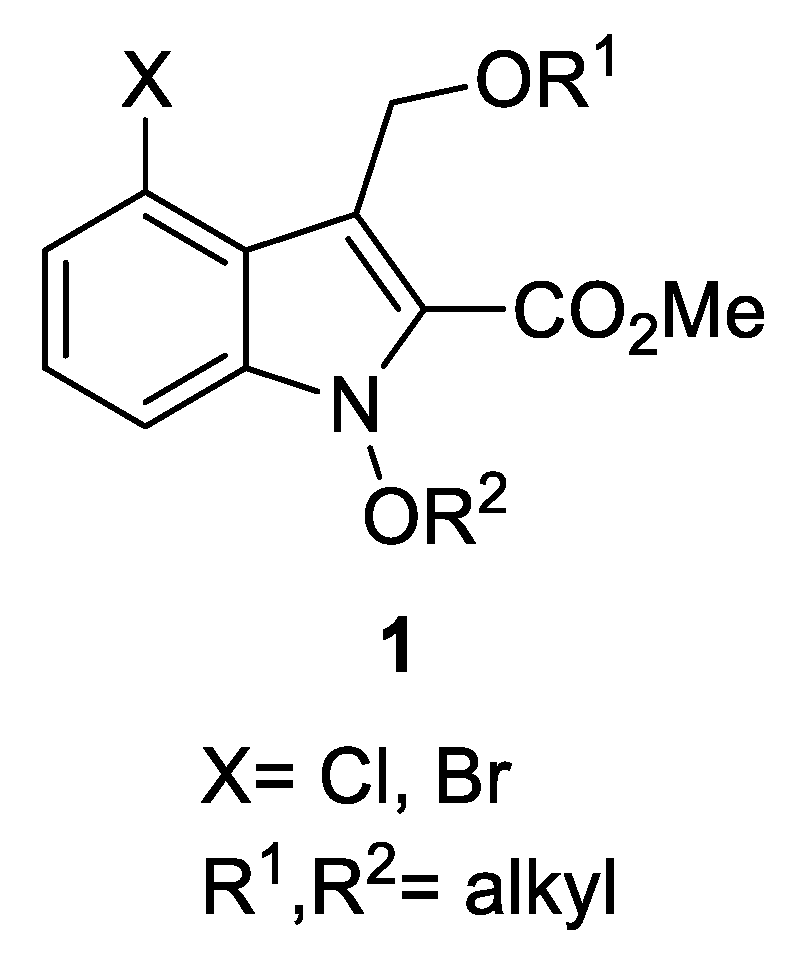
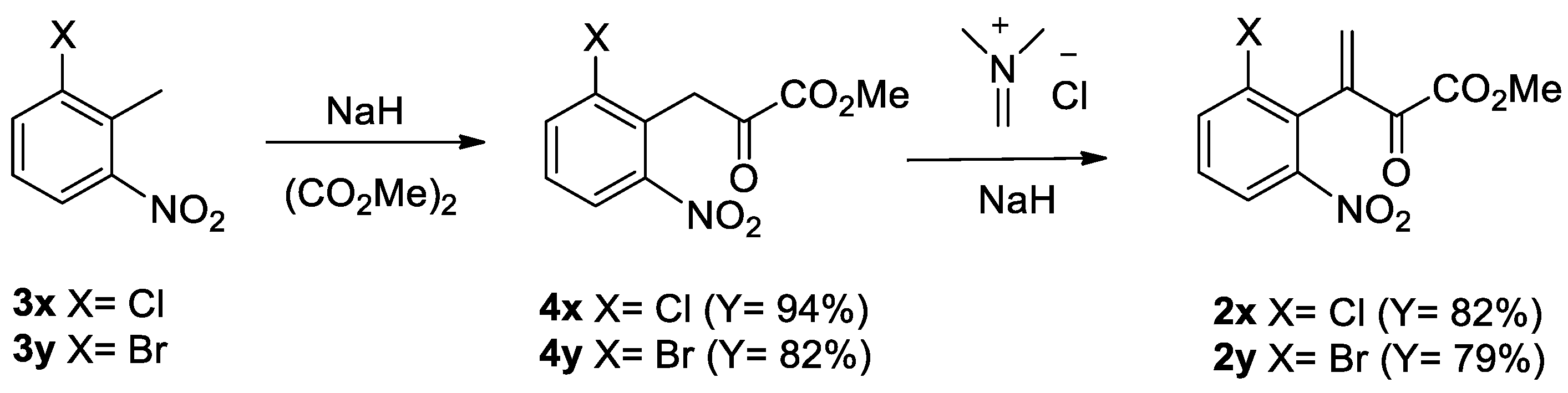
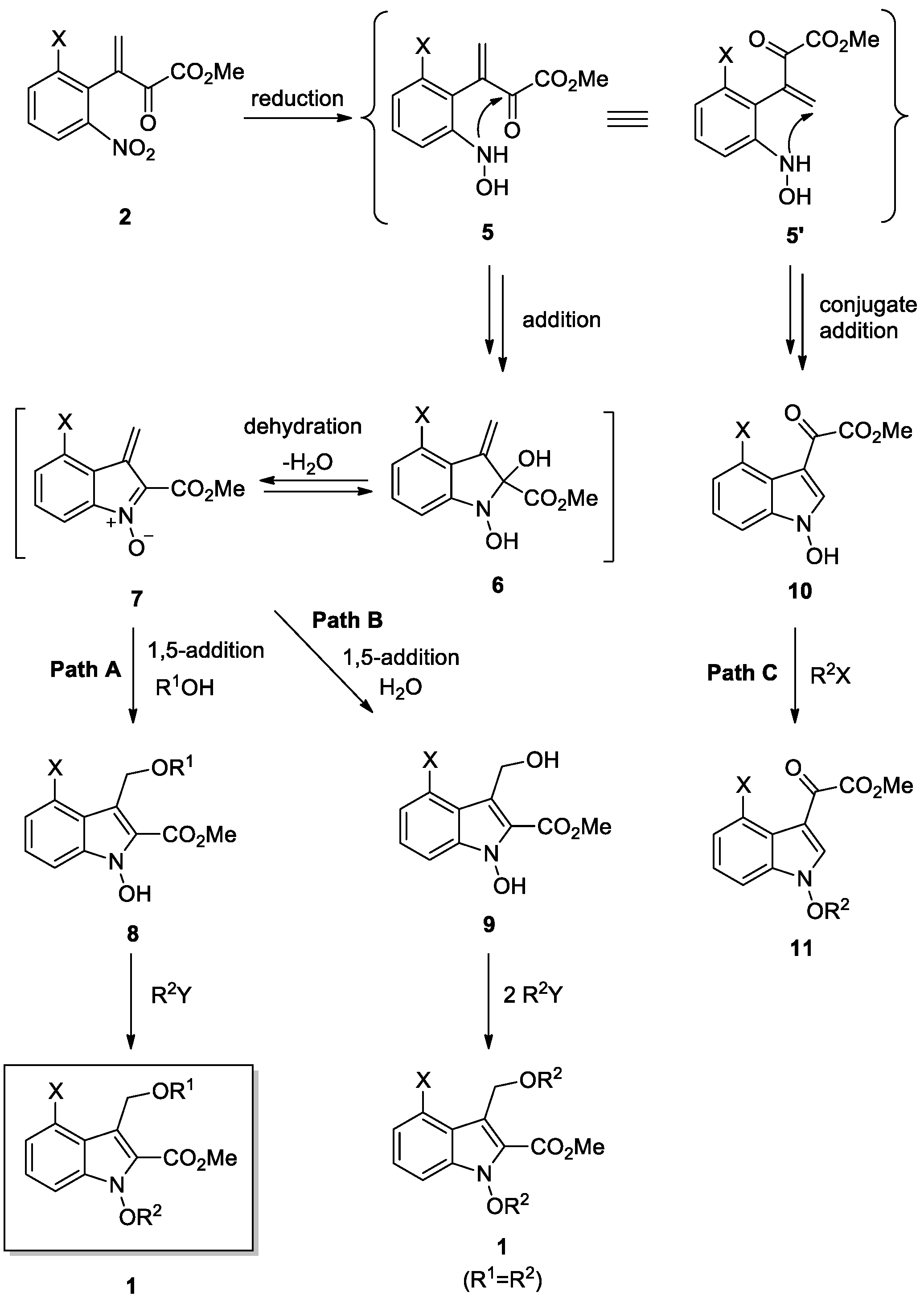
3.3. General Procedure for the Synthesis of 1-Alkoxyindoles 1
SnCl2·2H2O and 4Å molecular sieves stirred in DME for 30 min at 25 °C. To a stirred mixture was added alcohol and conjugate ketoester 2. The resulting mixture was stirred for 1–2 h at 40 °C. After checking that the starting material was disappeared by using TLC, DBU was added and stirred strongly for 30 min at 25 °C. The alkyl halide was then added and stirring was continued for 1–4 h at 25–50 °C until reaction completed. The reaction mixture was diluted with CH2Cl2 and washed with brine. The organic layer was dried (MgSO4) and concentrated in vacuo to afford a crude residue. The residue was purified by preparative TLC (PTLC) and column chromatography to give 1-alkoxyindoles 1. Spectral data of all compounds were in good accordance with the literature information.
Methyl 4-Chloro-1-methoxy-3-(methoxymethyl)-1H-indole-2-carboxylate (1xa)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2x (30 mg, 0.11 mmol, 1.0 eq) for 1 h at 40 °C then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and methyl iodide (14 μL, 0.22 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xa (13.7 mg, 43%) as a pale-yellow solid. Mp 59–60 °C; Rf 0.40 (1:4 EtOAc/hexanes); HPLC tR 20.8 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.38 (d, J = 8.2 Hz, 1H, Ar), 7.26 (t, J = 8.4 Hz, 1H, Ar), 7.18 (d, J = 5.9 Hz, 1H, Ar), 5.07 (s, 2H, C(3)CH2O), 4.18 (s, 3H, N(1)OCH3), 4.01 (s, 3H, CO2CH3), 3.45 (s, 3H, CH2OCH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 135.9 (Ar), 128.6 (Ar), 126.4 (Ar), 125.3 (Ar), 123.1 (Ar), 119.7 (Ar), 116.4 (Ar), 108.2 (Ar), 66.4 (N(1)OCH3), 63.7(CH2OCH3), 58.1 (C(3)CH2O), 52.4 (CO2CH3); MS m/z 283 [M]+; HRMS (+ESI) calcd for C13H14ClNNaO4 [M + Na]+ 306.0509, found 306.0509.
Methyl 4-Chloro-1-ethoxy-3-(ethoxymethyl)-1H-indole-2-carboxylate (1xb)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), ethanol (22 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and bromoethane (28 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xb (17.8 mg, 31%) as a pale-yellow solid. Mp 42 °C; Rf 0.49 (1:4 EtOAc/hexanes); HPLC tR 25.8 min; UV vis (CH3CN-H2O) λmax 235, 298nm; 1H NMR (300 MHz, CDCl3): δ 7.34 (d, J = 8.1 Hz, 1H, Ar), 7.23 (t, J = 7.8 Hz, 1H, Ar), 7.15 (d, J = 7.0 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.41 (q, J = 7.1 Hz, 2H, N(1)OCH2), 3.99 (s, 3H, CO2CH3), 3.65 (q, J = 7.0 Hz, 2H, OCH2CH3), 1.44 (t, J = 7.1 Hz, 3H, N(1)OCH2CH3), 1.25 (t, J = 7.0 Hz, 3H, OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.2 (Ar), 128.6 (Ar), 126.1 (Ar), 125.3 (Ar), 122.8 (Ar), 119.5 (Ar), 116.4 (Ar), 108.4 (Ar), 74.9 (N(1)OCH2), 65.8 (OCH2CH3), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 15.5 (N(1)OCH2CH3), 13.7 (OCH2CH3); MS m/z 311 [M]+; HRMS (+ESI) calcd for C15H18ClNNaO4 [M + Na]+ 334.0822, found 334.0820.
Methyl 4-Chloro-1-n-propyloxy-3-[(n-propyloxy)methyl]-1H-indole-2-carboxylate (1xc)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), n-propanol (28 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and 1-bromopropane (34 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xc (18.9 mg, 30%) as a white oil. Bp 170 °C (decomp.); Rf 0.54 (1:4 EtOAc/hexanes); HPLC tR 30.8 min; UV vis (CH3CN-H2O) λmax 230, 298nm; 1H NMR (300 MHz, CDCl3): δ 7.35 (d, J = 7.9 Hz, 1H, Ar), 7.23 (t, J = 8.0 Hz, 1H, Ar), 7.15 (d, J = 7.1 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.29 (t, J = 6.4 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.54 (t, J = 6.5 Hz, 2H, OCH2), 1.86 (sextet, J = 6.9 Hz, 2H, N(1)OCH2CH2), 1.64 (sextet, J = 7.0 Hz, 2H, OCH2CH2), 1.11 (t, J = 7.2 Hz, 3H, N(1)OCH2CH2CH3), 0.93 (t, J = 7.3 Hz, 3H, OCH2CH2CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.1 (Ar), 128.6 (Ar), 126.0 (Ar), 125.3(Ar), 122.8 (Ar), 119.6 (Ar), 116.4 (Ar), 108.3 (Ar), 80.6 (N(1)OCH2), 72.4 (OCH2), 62.1 (C(3)CH2O), 52.2 (CO2CH3), 23.1 (N(1)OCH2CH2), 21.8 (OCH2CH2), 10.9 (N(1)O(CH2)2CH3), 10.6 (O(CH2)2CH3); MS m/z 339 [M]+; HRMS (+ESI) calcd for C17H22ClNO4 [M + Na]+ 362.1135, found 362.1134.
Methyl 4-Chloro-1-n-butyloxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1xd)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromobutane (24 μL, 0.22 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xd (12.9 mg, 32%) as a white oil. Bp 198 °C (decomp.); Rf 0.54 (1:4 EtOAc/hexanes); HPLC tR 34.3 min; UV vis (CH3CN-H2O) λmax 235, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.34 (d, J = 8.2 Hz, 1H, Ar), 7.23 (t, J = 8.2 Hz, 1H, Ar), 7.15 (d, J = 7.4 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.5 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.58 (t, J = 6.5 Hz, 2H, OCH2), 1.82 (quintet, J = 7.0 Hz, 2H, N(1)OCH2CH2), 1.65–1.52 (m, 4H, OCH2CH2, N(1)OCH2CH2CH2), 1.39 (sextet, J = 7.5 Hz, 2H, O(CH2)2CH2), 1.00 (t, J = 7.4 Hz, 3H, N(1)O(CH2)3CH3), 0.90 (t, J = 7.4 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.1 (Ar), 128.6 (Ar), 126.0 (Ar), 125.3 (Ar), 122.8 (Ar), 119.6 (Ar), 116.4 (Ar), 108.3 (Ar), 79.0 (N(1)OCH2), 70.4 (OCH2), 62.1 (C(3)CH2O), 52.3 (CO2CH3), 32.1 (N(1)OCH2CH2), 30.5 (OCH2CH2) 19.6 (N(1)O(CH2)2CH2), 19.4 (O(CH2)2CH2) 14.2 (N(1)O(CH2)3CH3), 14.1 (O(CH2)3CH3); MS m/z 367 [M]+; HRMS (+ESI) calcd for C19H26ClNO4 [M + Na]+ 390.1448, found 390.1447.
Methyl 4-Chloro-1-n-pentyloxy-3-[(n-pentyloxy)methyl]-1H-indole-2-carboxylate (1xe)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-pentanol (24 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromopentane (28 μL, 0.22 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xe (8.7 mg, 20%) as a white oil. Bp 184 °C (decomp.); Rf 0.58 (1:4 EtOAc/hexanes); HPLC tR 32.9 min; UV vis (CH3CN-H2O) λmax 235, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.34 (d, J = 8.1 Hz, 1H, Ar), 7.23 (t, J = 8.1 Hz, 1H, Ar), 7.15 (d, J = 7.5 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.59 (t, J = 6.7 Hz, 2H, OCH2), 1.84 (quintet, J = 7.4 Hz, 2H, N(1)OCH2CH2), 1.64–1.25 (m, 10H, OCH2(CH2)3CH3, N(1)OCH2CH2(CH2)2CH3), 0.95 (t, J = 7.2 Hz, 3H, N(1)O(CH2)4CH3), 0.87 (t, J = 6.9 Hz, 3H, O(CH2)4CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.0 (Ar), 128.6 (Ar), 126.0 (Ar), 125.3 (Ar), 122.8 (Ar), 119.6 (Ar), 116.4 (Ar), 108.2 (Ar), 79.3 (N(1)OCH2), 70.7 (OCH2), 62.1 (C(3)CH2O), 52.2 (CO2CH3), 29.7, 28.6, 28.3, 28.1, 22.7, 22.6, (N(1)OCH2(CH2)3, OCH2(CH2)3), 14.2 (N(1)O(CH2)4CH3), 14.1 (O(CH2)4CH3); MS m/z 395 [M]+; HRMS (+ESI) calcd for C21H30ClNO4 [M + Na]+ 418.1761, found 418.1760.
Methyl 4-Chloro-1-n-hexyloxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1xf)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), n-hexanol (74 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and 1-bromohexane (52 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xf (16.5 mg, 21%) as a pale-yellow oil. Bp 152 °C (decomp.); Rf 0.63 (1:4 EtOAc/hexanes); HPLC tR 29.7 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.33 (d, J = 8.2 Hz, 1H, Ar), 7.23 (t, J = 8.0 Hz, 1H, Ar), 7.14 (d, J = 8.0 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, OCH3), 3.57 (t, J = 6.6 Hz, 2H, OCH2), 1.83 (quintet, J = 6.7 Hz, 2H, N(1)OCH2CH2), 1.63–1.50 (m, 4H, OCH2CH2, N(1)O(CH2)2CH2), 1.37–1.25 (m, 10H, OCH2CH2(CH2)3, N(1)OCH2CH2CH2(CH2)2), 0.92–0.84 (m, 6H, N(1)O(CH2)5CH3, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3): δ 160.7 (C=O), 135.9 (Ar), 128.5 (Ar), 125.8 (Ar), 125.1(Ar), 122.6 (Ar), 119.4 (Ar), 116.2 (Ar), 108.1 (Ar), 79.1 (N(1)OCH2), 70.5 (OCH2), 61.9 (C(3)CH2O), 52.0 (CO2CH3), 31.7, 31.6, 29.8, 28.2, 25.9, 25.6, 22.6, 22.5 (N(1)OCH2(CH2)4, OCH2(CH2)4), 14.1 (N(1)O(CH2)5CH3), 14.0 (O(CH2)5CH3); MS m/z 423 [M]+; HRMS (+ESI) calcd for C23H34ClNO4 [M + Na]+ 446.2074, found 446.2071.
Methyl 4-Chloro-1-n-octyloxy-3-[(n-octyloxy)methyl]-1H-indole-2-carboxylate (1xg)
Use of SnCl2·2H2O (166 mg, 0.74 mmol, 3.3 eq), n-octanol (70 μL, 0.45 mmol, 2.0 eq) and 2x (60 mg, 0.22 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (350 μL, 2.20 mmol, 10.0 eq) and 1-bromooctane (80 μL, 0.45 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xg (23.2 mg, 22%) as a white solid. Mp 19–20 °C; Rf 0.70 (1:4 EtOAc/hexanes); HPLC tR 35.5 min; UV vis (CH3CN-H2O) λmax 237, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.33 (d, J = 8.2 Hz, 1H, Ar), 7.23 (t, J = 8.1 Hz, 1H, Ar), 7.14 (d, J = 7.4 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.56 (t, J = 6.7 Hz, 2H, OCH2), 1.83 (quintet, J = 7.0 Hz, 2H, N(1)OCH2CH2), 1.66–1.25 (m, 22H, OCH2(CH2)6CH3), N(1)OCH2CH2(CH2)5CH3), 0.89–0.85 (m, 6H, N(1)O(CH2)7CH3, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.1 (Ar), 128.7 (Ar), 126.0 (Ar), 125.3(Ar), 122.8 (Ar), 120.0 (Ar), 116.4 (Ar), 108.3(Ar), 79.3 (N(1)OCH2), 70.7 (OCH2), 62.1 (C(3)CH2O), 52.2 (CO2CH3), 32.0, 31.9, 30.0, 29.9, 29.6, 29.5, 29.4, 28.5, 26.4, 26.2, 22.8, 22.6 5 (N(1)CH2(CH2)6, OCH2(CH2)6), 14.3 (N(1)O(CH2)7CH3), 13.5 (O(CH2)7CH3); MS m/z 479 [M]+; HRMS (+ESI) calcd for C27H42ClNNaO4 [M + Na]+ 502.2700, found 502.2926.
Methyl 4-Chloro-1-benzyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1xh)
Use of SnCl2·2H2O (38.4 mg, 0.17 mmol, 3.3 eq), benzyl alcohol (12 μL, 0.11 mmol, 2.0 eq) and 2x (14.5 mg, 0.05 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (165 μL, 0.55 mmol, 10.0 eq) and benzyl bromide (14 μL, 0.11 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xh (7.4 mg, 32%) as a white solid. Mp 74 °C; Rf 0.54 (1:2 EtOAc/hexanes); HPLC tR 31.8 min; UV vis (CH3CN-H2O) λmax 212, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.50–7.10 (m, 13H, Ar), 5.32 (s, 2H, C(3)CH2O), 5.17 (s, 2H, N(1)OCH2), 4.66 (s, 2H, C(3)CH2OCH2), 3.87 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 138.8 (Ar), 136.4 (Ar), 134.3 (Ar), 130.0 (Ar), 129.5 (Ar), 128.9 (Ar), 128.6 (Ar), 128.5 (Ar), 128.2 (Ar), 127.7 (Ar), 126.2 (Ar), 125.9 (Ar), 123.0 (Ar), 119.7 (Ar), 116.2 (Ar), 108.6 (Ar), 81.0 (N(1)OCH2), 72.6 (OCH2Ph), 61.8 (C(3)CH2O), 52.2 (CO2CH3); MS m/z 435 [M]+; HRMS (+ESI) calcd for C25H22ClNNaO4 [M + Na]+ 458.1135, found 458.1133.
Methyl 4-Chloro-1-phenylethyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1xi)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), 2-phenylethyl alcohol (46 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and 2-(bromoethyl)benzene (51 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xi (15.2 mg, 18%) as a pale yellow solid. Mp 50 °C; Rf 0.60 (1:2 EtOAc/hexanes); HPLC tR 34.5 min; UV vis (CH3CN-H2O) λmax 212, 236, 297 nm; 1H NMR (300 MHz, CDCl3): δ 7.38–6.90 (m, 13H, Ar), 5.14 (s, 2H, C(3)CH2O), 4.55 (t, J = 6.7 Hz, 2H, N(1)OCH2), 3.86 (s, 3H, CO2CH3), 3.79 (t, J = 7.4 Hz, 2H, C(3)CH2OCH2), 3.14 (t, J = 6.7 Hz, 2H, N(1)OCH2CH2), 2.94 (t, J = 7.5 Hz, 2H, C(3)CH2OCH2CH2); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 139.2 (Ar), 137.8 (Ar), 136.2 (Ar), 129.3 (Ar), 129.1 (Ar), 128.8 (Ar), 128.5 (Ar), 128.4 (Ar), 126.9 (Ar), 126.2 (Ar), 125.9 (Ar), 125.4 (Ar), 122.9 (Ar), 119.6 (Ar), 116.4 (Ar), 108.3 (Ar), 79.6 (N(1)OCH2), 71.4 (OCH2), 62.2 (C(3)CH2O), 52.2 (CO2CH3), 36.5 (N(1)OCH2CH2), 35.0 (OCH2CH2); MS m/z 463 [M]+; HRMS (+ESI) calcd for C27H26ClNNaO4 [M + Na]+ 486.1448, found 486.1445.
Methyl 4-Chloro-1-isopropyloxy-3-[(isopropyloxy)methyl]-1H-indole-2-carboxylate (1xj)
Use of SnCl2·2H2O (166 mg, 0.74 mmol, 3.3 eq), isopropanol (35 μL, 0.45 mmol, 2.0 eq) and 2x (60 mg, 0.22 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (350 μL, 2.20 mmol, 10.0 eq) and 2-bromopropane (43 μL, 0.45 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xj (9.2 mg, 12%) as a white oil. Bp 164 °C (decomp.); Rf 0.50 (1:4 EtOAc/hexanes); HPLC tR 29.1 min; UV vis (CH3CN-H2O) λmax 233, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.35 (d, J = 8.2 Hz, 1H, Ar), 7.20 (t, J = 7.6 Hz, 1H, Ar), 7.13 (d, J = 7.4 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.72 (septet, J = 6.2 Hz, 1H, N(1)OCH(CH3)2), 3.80 (septet, 1H, C(3)CH2OCH(CH3)2), 1.33 (d, J = 6.2 Hz, 6H, N(1)OCH(CH3)2), 1.24 (d, J = 6.1 Hz, 6H, C(3)CH2OCH(CH3)2); 13C NMR (75 MHz, CDCl3): δ 161.1 (C=O), 137.5 (Ar), 128.4 (Ar), 126.1 (Ar), 125.8 (Ar), 122.6 (Ar), 119.5 (Ar), 116.8 (Ar), 109.4 (Ar), 82.1 (N(1)OCH), 71.5 (OCH), 60.0 (C(3)CH2O), 52.2 (CO2CH3), 22.4 (N(1)OCH(CH3)2), 21.3 (OCH(CH3)2); MS m/z 339 [M]+; HRMS (+ESI) calcd for C17H22ClNNaO4 [M + Na]+ 362.1135, found 362.1131.
Methyl 4-Chloro-1-cyclohexyloxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1xk)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and bromocyclohexane (27 μL, 0.22 mmol, 2.0 eq) for 4 h at 50 °C in general procedure afforded the title compound 1xk (4.7 mg, 11%) as a white solid. Mp 60–64 °C; Rf 0.57 (1:4 EtOAc/hexanes); HPLC tR 31.9 min; UV vis (CH3CN-H2O) λmax 237, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.37 (d, J = 8.2 Hz, 1H, Ar), 7.20 (t, J = 7.6 Hz, 1H, Ar), 7.12 (d, J = 7.5 Hz, 1H, Ar), 5.11 (s, 2H, C(3)CH2O), 4.35–4.28 (m, 1H, N(1)OCH), 3.97 (s, 3H, CO2CH3), 3.45–3.38 (m, 1H, C(3)CH2OCH), 2.38–0.86 (m, 20H, N(1)OCH(CH2)5, C(3)CH2OCH(CH2)5); 13C NMR (75 MHz, CDCl3): δ 161.1 (C=O), 137.3 (Ar) 128.4 (Ar), 126.0 (Ar), 125.7 (Ar), 122.5 (Ar), 119.2 (Ar), 116.7 (Ar), 109.4 (Ar), 87.9 (N(1)OCH), 78.3 (C(3)CH2OCH), 59.7 (C(3)CH2O), 52.2 (CO2CH3), 31.7, 29.9. 26.1, 25.6, 24.7, 24.6 (N(1)OCH(CH2)5, OCH(CH2)5); MS m/z 419 [M]+; HRMS (+ESI) calcd for C23H30ClNNaO4 [M + Na]+ 442.1760, found 442.1760.
Methyl 4-Chloro-1-methoxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1xl)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), 2-phenylethyl alcohol (46 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and methyl iodide (23 μL, 0.37 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xl (15.2 mg, 22%) as a white solid. Mp 68 °C; Rf 0.30 (1:4 EtOAc/hexanes); HPLC tR 29.0 min; UV vis (CH3CN-H2O) λmax 215, 234, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.38 (d, J = 8.2 Hz, 2H, Ar), 7.29–7.16 (m, 6H, Ar), 5.15 (s, 2H, C(3)CH2O,), 4.19 (s, 3H, N(1)OCH3), 3.96 (s, 3H, CO2CH3), 3.81 (t, J = 7.4 Hz, 2H, OCH2), 2.95 (t, J = 7.4 Hz, 2H, OCH2CH2); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 139.3 (Ar), 135.9 (Ar), 129.1 (Ar), 128.7 (Ar), 128.5 (Ar), 126.3 (Ar), 126.2 (Ar), 125.3 (Ar), 123.0 (Ar), 119.7 (Ar), 116.5 (Ar), 108.2 (Ar), 71.5 (N(1)OCH3), 66.4 (OCH2), 62.2 (C(3)CH2O), 52.4 (CO2CH3), 36.5 (OCH2CH2); MS m/z 373 [M]+; HRMS (+ESI) calcd for C20H20ClNNaO4 [M + Na]+ 396.0979, found 396.0977.
Methyl 4-Chloro-1-n-octyloxy -3-[(methoxymethyl]-1H-indole-2-carboxylate (1xm)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromooctane (38 μL, 0.22 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xm (13.7 mg, 33%) as a yellow oil. Bp 178 °C (decomp.); Rf 0.67 (1:2 EtOAc/hexanes); HPLC tR 26.3 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.36 (dd, J = 8.2, 1.0 Hz, 1H, Ar), 7.25 (t, J = 7.8 Hz, 1H, Ar), 7.16 (dd, J = 7.5, 1.0 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O,), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 4.00 (s, 3H, CO2CH3), 3.46 (s, 3H, OCH3), 1.83 (quintet, J = 7.1 Hz, 2H, OCH2CH2), 1.54–1.47 (m, 2H, O(CH2)2CH2), 1.34–1.25 (m, 8H, O(CH2)3(CH2)4, 0.90 (t, J = 7.0 Hz, 3H, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.0 (Ar), 128.5 (Ar), 126.1 (Ar), 125.2 (Ar), 122.9 (Ar), 119.5 (Ar), 116.1 (Ar), 108.4 (Ar), 79.4 (N(1)OCH2), 63.7 (OCH3), 58.1 (C(3)CH2O), 52.3 (CO2CH3), 32.0, 29.6, 29.4, 28.4, 26.2, 22.8 (N(1)OCH2(CH2)6), 14.3 N(1)O(CH2)7CH3; MS m/z 381 [M]+; HRMS (+ESI) calcd for C20H28ClNO4 [M]+ 381.1707, found 381.1707.
Methyl 4-Chloro-1-methoxy-3-[(n-octyloxy)methyl]-1H-indole-2-carboxylate (1xn)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-octanol (35 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and methyl iodide (14 μL, 0.22 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xn (15.0 mg, 36%) as a white solid. Mp 36 °C; Rf 0.74 (1:2 EtOAc/hexanes); HPLC tR 27.0 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.37 (d, J = 8.2 Hz, 1H, Ar), 7.26 (t, J = 7.8 Hz, 1H, Ar), 7.16 (d, J = 7.1 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O,), 4.19 (s, 3H, N(1)OCH3), 4.00 (s, 3H, CO2CH3), 3.57 (t, J = 6.7 Hz, 2H, OCH2), 1.66–1.59 (m, 2H, OCH2CH2), 1.43–1.25 (m, 10H, O(CH2)2(CH2)5), 0.87 (t, J = 6.6 Hz, 3H, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.0 (Ar), 128.8 (Ar), 126.3 (Ar), 125.3 (Ar), 122.9 (Ar), 119.7 (Ar), 116.7 (Ar), 108.1 (Ar), 70.8 (N(1)OCH3), 66.4 (OCH2), 62.1 (C(3)CH2O), 52.4 (CO2CH3), 32.0, 30.0, 29.6, 29.5, 26.4, 22.9 (OCH2(CH2)6), 14.3 (O(CH2)7CH3); MS m/z 381 [M]+; HRMS (+ESI) calcd for C20H28ClNO4 [M]+ 381.1707, found 381.1707.
Methyl 4-Bromo-1-methoxy-3-(methoxymethyl)-1H-indole-2-carboxylate (1ya)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), methanol (8 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and methyl iodide (13 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1ya (17.1 mg, 52%) as a white solid. Mp 50–52 °C; Rf 0.29 (1:4 EtOAc/hexanes); HPLC tR 34.3 min; UV vis (CH3CN-H2O) λmax 215, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.49–7.38 (m, 2H, Ar), 7.19 (t, J = 8.1 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.18 (s, 3H, N(1)OCH3), 4.01 (s, 3H, CO2CH3), 3.47 (s, 3H, CH2OCH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 135.8 (Ar), 131.2 (Ar), 126.7 (Ar), 125.6 (Ar), 123.8 (Ar), 120.9 (Ar), 116.7 (Ar), 108.8 (Ar), 66.4 (N(1)OCH3), 63.0 (CH2OCH3), 58.0 (C(3)CH2O), 52.4 (CO2CH3); MS m/z 327 [M]+; HRMS (+ESI) calcd for C13H14BrNNaO4 [M + Na]+ 350.0004, found 350.0002.
Methyl 4-Bromo-1-ethoxy-3-(ethoxymethyl)-1H-indole-2-carboxylate (1yb)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), ethanol (12 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and bromoethane (16 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yb (9.3 mg, 26%) as a white solid. Mp 42 °C; Rf 0.51 (1:2 EtOAc/hexanes); HPLC tR 26.2 min; UV vis (CH3CN-H2O) λmax 235, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.45–7.33 (m, 2H, Ar), 7.16 (t, J = 7.9 Hz, 1H, Ar), 5.10 (s, 2H, C(3)CH2O), 4.41 (q, J = 7.1 Hz, 2H, N(1)OCH2), 3.99 (s, 3H, CO2CH3), 3.66 (q, J = 7.0 Hz, 2H, OCH2), 1.44 (t, J = 7.1 Hz, 3H, N(1)OCH2CH3), 1.26 (t, J = 7.0 Hz 3H, OCH2CH3), 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.2 (Ar), 126.5 (Ar), 126.3 (Ar), 120.8 (Ar), 125.5(Ar), 116.7 (Ar), 116.4 (Ar), 109.0 (Ar), 75.0 (N(1)OCH2), 65.8 (OCH2), 61.4 (C(3)CH2O), 52.3 (CO2CH3), 15.6 (N(1)OCH2CH3), 13.8 (OCH2CH3); MS m/z 355 [M]+; HRMS (+ESI) calcd for C15H19BrNNaO4 [M + Na]+ 378.0317, found 378.0315.
Methyl 4-Bromo-1-n-propyloxy-3-[(n-propyloxy)methyl]-1H-indole-2-carboxylate (1yc)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), n-propanol (15 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and 1-bromopropane (18 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yc (10.9 mg, 28%) as a white oil. Bp 224 °C (decomp.); Rf 0.62 (1:2 EtOAc/hexanes); HPLC tR 31.3 min; UV vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.40 (d, J = 8.3 Hz, 1H, Ar), 7.39 (d, J = 8.3 Hz, 1H, Ar), 7.15 (t, J = 7.7 Hz, 1H, Ar), 5.10 (s, 2H, C(3)CH2O), 4.29 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.55 (t, J = 6.6 Hz, 2H, OCH2), 1.86 (sextet, J = 7.2 Hz, 2H, N(1)OCH2CH2), 1.66 (sextet, J = 7.2 Hz, 2H, OCH2CH2), 1.11 (t, J = 7.4 Hz, 3H, N(1)OCH2CH2CH3), 0.94 (t, J = 7.4 Hz, 3H, OCH2CH2CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.0 (Ar), 126.5 (Ar), 126.3 (Ar), 125.7(Ar), 120.9 (Ar), 116.8 (Ar), 116.4 (Ar), 108.9 (Ar), 80.7 (N(1)OCH2), 72.3 (OCH2), 61.5 (C(3)CH2O), 52.3 (CO2CH3), 23.2 (N(1)OCH2CH2), 21.8 (OCH2CH2), 11.0 (N(1)O(CH2)2CH3), 10.7 (O(CH2)2CH3); MS m/z 383 [M]+; HRMS (+ESI) calcd for C17H22BrNNaO4 [M + Na]+ 406.0630, found 408.0608.
Methyl 4-Bromo-1-n-octyloxy-3-[(n-octyloxy)methyl]-1H-indole-2-carboxylate (1yg)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), n-octanol (9 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and 1-bromooctane (35 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yg (11.0 mg, 21%) as a yellow oil. Bp 194 °C (decomp.); Rf 0.40 (1:10 EtOAc/hexanes); HPLC tR 36.1 min; UV vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.37 (t, J = 7.9 Hz, 2H, Ar), 7.16 (t, J = 7.9 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.57 (t, J = 6.6 Hz, 2H, OCH2), 1.83 (quintet, J = 7.5 Hz, 2H, N(1)OCH2CH2), 1.66–1.15 (m, 22H, N(1)OCH2CH2(CH2)5, OCH2(CH2)6), 1.00–0.77 (m, 6H, N(1)O(CH2)7CH3, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.0 (Ar), 126.4 (Ar), 126.3 (Ar), 125.6 (Ar), 120.9 (Ar), 116.8 (Ar), 116.4 (Ar), 108.9(Ar), 79.4 (N(1)OCH2), 70.6 (OCH2), 61.5 (C(3)CH2O), 53.3 (CO2CH3), 53.3, 52.3, 32.0, 30.1, 30.0, 29.7, 29.5, 29.4, 28.5, 26.5, 26.2, 22.9 (N(1)OCH2(CH2)6,OCH2(CH2)6), 14.3 (N(1)O(CH2)7CH3), 13.5 (O(CH2)7CH3); MS m/z 523 [M]+; HRMS (+ESI) calcd for C27H42BrNNaO4 [M + Na]+ 546.2195, found 546.2190.
Methyl 4-Bromo-1-benzyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1yh)
Use of SnCl2·2H2O (38 mg, 0.17 mmol, 3.3 eq), benzyl alcohol (11 μL, 0.10 mmol, 2.0 eq) and 2y (15.7 mg, 0.05 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (75 μL, 0.50 mmol, 10.0 eq) and benzyl bromide (12 μL, 0.10 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yh (9.8 mg, 41%) as a pale-yellow solid. Mp 84 °C; Rf 0.29 (1:2 EtOAc/hexanes); HPLC tR 33.3 min; UV vis (CH3CN-H2O) λmax 228, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.55–7.22 (m, 12H, Ar), 7.13 (t, J = 7.9 Hz, 1H, Ar), 5.32 (s, 2H, C(3)CH2O), 5.19 (s, 2H, N(1)OCH2), 4.68 (s, 2H, OCH2), 3.88 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 138.8 (Ar), 136.3 (Ar), 134.3 (Ar), 130.0 (Ar), 129.5 (Ar), 129.0 (Ar), 128.5 (Ar), 128.3 (Ar), 127.7 (Ar), 126.7 (Ar), 126.4 (Ar), 126.2 (Ar), 121.0 (Ar), 116.5 (Ar), 116.3 (Ar) 109.2 (Ar), 81.0 (N(1)OCH2), 72.6 (OCH2Ph), 61.2(C(3)CH2O), 52.3 (CO2CH3); MS m/z 479 [M]+; HRMS (+ESI) calcd for C25H22BrNNaO4 [M + Na]+ 502.0630, found 502.0626.
Methyl 4-Bromo-1-isopropyloxy-3-[(isopropyloxy)methyl]-1H-indole-2-carboxylate (1yj)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), isopropanol (15 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and 2-bromopropane (19 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yj (6.2 mg, 16%) as a white oil. Bp 178 °C (decomp.); Rf 0.69 (1:2 EtOAc/hexanes); HPLC tR 30.5 min; UV vis (CH3CN-H2O) λmax 235, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.40 (d, J = 8.3 Hz, 1H, Ar), 7.35 (d, J = 7.5 Hz, 1H, Ar), 7.13 (t, J = 7.9 Hz, 1H, Ar), 5.11 (s, 2H, C(3)CH2O), 4.72 (septet, J = 6.2 Hz, 1H, N(1)OCH(CH3)2), 3.97 (s, 3H, CO2CH3), 3.82 (septet, J = 6.0 Hz, 1H, C(3)CH2OCH(CH3)2), 1.34 (d, J = 6.2 Hz, 6H, N(1)OCH(CH3)2), 1.26 (d, J = 6.1 Hz, 6H, OCH(CH3)2); 13C NMR (75 MHz, CDCl3): δ 161.1 (C=O), 137.3 (Ar) 126.3 (Ar), 126.1 (Ar), 120.7 (Ar), 117.1 (Ar), 116.1 (Ar), 109.9 (Ar), (one Ar peak was not detected and believed to overlap with the observed peak), 82.2 (N(1)OCH(CH3)2), 71.5 (OCH(CH3)2), 59.4 (C(3)OCH2), 52.2 (CO2CH3), 22.4 (N(1)OCH(CH3)2), 21.3 (OCH(CH3)2); MS m/z 383 [M]+; HRMS (+ESI) calcd for C17H22BrNNaO4 [M + Na]+ 406.0630, found 406.0632.
Methyl 4-Bromo-1-methoxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1yl)
Use of SnCl2·2H2O (37 mg, 0.17 mmol, 3.3 eq), 2-phenylethyl alcohol (12 μL, 0.10 mmol, 2.0 eq) and 2y (15.7 mg, 0.05 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (75 μL, 0.50 mmol, 10.0 eq) and methyl iodide (4 μL, 0.10 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yl (5.2 mg, 25%) as a white solid. Mp 59 °C; Rf 0.57 (1:2 EtOAc/hexanes); HPLC tR 29.7 min; UV vis (CH3CN-H2O) λmax 235, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.43 (d, J = 8.3 Hz, 1H, Ar), 7.38 (d, J = 7.4 Hz, 1H, Ar), 7.29–7.16 (m, 6H, Ar), 5.15 (s, 2H, C(3)CH2O), 4.19 (s, 3H, N(1)OCH3), 3.96 (s, 3H, CO2CH3), 3.82 (t, J = 7.4 Hz, 2H, OCH2), 2.96 (t, J = 7.4 Hz, 2H, OCH2CH2); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 139.3 (Ar), 135.9 (Ar), 129.1 (Ar), 128.5 (Ar), 126.7 (Ar), 126.5 (Ar), 126.3 (Ar), 125.6 (Ar), 120.9 (Ar), 116.8 (Ar), 116.4 (Ar), 108.8 (Ar), 71.5 (N(1)OCH3), 66.4 (OCH2), 61.6 (C(3)CH2O), 52.4 (CO2CH3), 36.5 (OCH2CH2); MS m/z 419 [M]+; HRMS (+ESI) calcd for C20H20BrNNaO4 [M + Na]+ 440.0473, found 440.0471.
Methyl 4-Bromo-1-pentyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ym)
Use of SnCl2·2H2O (37 mg, 0.17 mmol, 3.3 eq), 2-phenylethyl alcohol (12 μL, 0.10 mmol, 2.0 eq), and 2y (15.7 mg, 0.05 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (75 μL, 0.50 mmol, 10.0 eq) and 1-bromopentane (13 μL, 0.10 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1ym (5.0 mg, 21%) as a white oil. Bp 224 °C (decomp.); Rf 0.70 (1:2 EtOAc/hexanes); HPLC tR 36.7 min; UV vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CD3CN): δ 7.38 (t, J = 8.1 Hz, 2H, Ar), 7.30–7.10 (m, 6H, Ar), 5.16 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.95 (s, 3H, CO2CH3), 3.81 (t, J = 7.5 Hz, 2H, OCH2CH2Ph), 2.96 (t, J = 7.4 Hz, 2H, OCH2CH2Ph), 1.95–1.73 (m, 2H, N(1)OCH2CH2), 1.58–1.30 (m, 4H, N(1)OCH2CH2(CH2)2), 0.96 (t, J = 7.1 Hz, 3H, N(1)O(CH2)4CH3); 13C NMR (75 MHz, CD3CN): δ 160.7 (C=O), 139.3 (Ar), 136.0 (Ar), 129.1 (Ar), 128.5 (Ar), 126.5 (Ar), 126.4 (Ar), 126.3 (Ar), 125.6 (Ar), 120.8 (Ar), 116.5 (Ar), 116.4 (Ar), 108.9 (Ar), 79.4 (N(1)OCH2), 71.4 (OCH2CH2Ph), 61.6 (C(3)CH2O), 52.3 (CO2CH3), 36.6 (OCH2CH2Ph), 28.3 (N(1)OCH2CH2), 28.2 (N(1)O(CH2)2CH2), 22.8 (N(1)O(CH2)3CH2), 14.2 (N(1)O(CH2)4CH3); MS m/z 473 [M]+; HRMS (+ESI) calcd for C24H28BrNNaO4 [M+Na]+ 496.1099, found 495.1097.
Methyl 2-(4′-Chloro-1′-hydroxy-1′H-indol-3′-yl)-2-oxoacetate (10x) [19] Brown solid. Mp 84–86 °C; Rf 0.15 (2:1 EtOAc/hexanes); 1H NMR (300 MHz, CD3CN): δ 9.45 (br s, 1H, OH), 8.27 (s, 1H), 7.51 (dd, J = 5.0, 1.0 Hz, 1H), 7.40 (t, J = 4.7 Hz, 1H), 7.33 (dd, J = 4.2, 1.0 Hz, 1H), 3.90 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 179.5, 164.3, 137.5, 127.2, 126.5, 126.4, 126.1, 117.4, 110.0, 109.5, 53.8; MS m/z 276 [M + Na]+; HRMS (+ESI) Calcd for C11H8ClNNaO4 [M + Na]+ 276.0040, found 276.0034.
Methyl 2-(4′-Chloro-1′-methoxy-1′H-indol-3′-yl)-2-oxoacetate (11xl)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), 2-phenylethyl alcohol (46 μL, 0.37 mmol, 2.0 eq), and 2x (50 mg, 0.185 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and methyl iodide (23 μL, 0.37 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the compound 11xl (15.8 mg, 32%) as a brown solid. Mp 64 °C; Rf 0.50 (2:1 EtOAc/hexanes); HPLC tR 16.6 min; UV vis (CH3CN-H2O) λmax 218, 262, 321 nm; 1H NMR (300 MHz, CDCl3): δ 8.40 (s, 1H, C(2)H), 7.43–7.15 (m, 3H, Ar), 4.19 (s, 3H, N(1)OCH3), 3.95 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3): δ 177.7 (C(3)C=O), 164.1 (C=O), 134.3 (Ar), 133.5 (Ar), 128.1 (Ar), 125.4 (Ar), 125.3 (Ar), 120.7 (Ar), 109.6 (Ar), 107.8 (Ar), 67.6 (N(1)OCH3), 53.2 (CO2CH3); MS m/z 267 [M]+; HRMS (+ESI) calcd for C12H10ClNNaO4 [M + Na]+ 290.0196, found 290.0193.
Methyl 2-(4′-Chloro-1′-octyloxy-1′H-indol-3′-yl)-2-oxoacetate (11xm)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromooctane (38 μL, 0.22 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the compound 11xm (1.1 mg, 3%) as a brown solid. Mp 40 °C; Rf 0.57 (1:2 EtOAc/hexanes); HPLC tR 33.4 min; UV vis (CH3CN-H2O) λmax 219, 262, 320 nm; 1H NMR (300 MHz, CDCl3): δ 8.37 (s, 1H, C(2)H), 7.40–7.15 (m, 3H, Ar), 4.31 (t, J = 6.7 Hz, 2H, N(1)OCH2), 3.95 (s, 3H, OCH3), 1.82 (quintet, J = 7.1 Hz, 2H, N(1)OCH2CH2), 1.57–1.25 (m, 10H, O(CH2)2(CH2)5, 0.90 (t, J = 6.6 Hz, 3H, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 177.6 (C(3)C=O), 164.2 (C=O), 134.9 (Ar), 134.2 (Ar), 128.1 (Ar), 125.4 (Ar), 125.2 (Ar), 109.4 (Ar), 108.0 (Ar), (one Ar peak was not detected and believed to overlap with the observed peak), 80.5 (N(1)OCH2), 53.1 (CO2CH3), 32.1, 29.9, 29.5, 28.3, 25.9, 22.8 (N(1)OCH2(CH2)6), 14.3 (N(1)O(CH2)7CH3); MS m/z 365 [M]+; HRMS (+ESI) calcd for C19H24ClNO4 [M]+ 365.1394, found 365.1394.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


 1xa
1xa 1xb
1xb 1xc
1xc 1xd
1xd 1xe
1xe 1xf
1xf 1xg
1xg 1xh
1xh 1xi
1xi 1xj
1xj 1xk
1xk 1xl
1xl 1xm
1xm 1xn
1xn 1ya
1ya 1yb
1yb 1yc
1yc 1yg
1yg 1yh
1yh 1yj
1yj 1yl
1yl 1ym
1ym