
Lipid Formulations and Bioconjugation Strategies for Indomethacin Therapeutic Advances



Abstract
:1. Introduction
2. Therapeutic Activity of Indomethacin and Its Mechanism of Action
3. Side Effects of Indomethacin and Other NSAIDs
4. Novel Strategies for Indomethacin Delivery
4.1. Associations of Phospholipids with Indomethacin and Lipid Emulsions of Indomethacin
4.2. Liposomal Formulations of Indomethacin and Their Efficacy
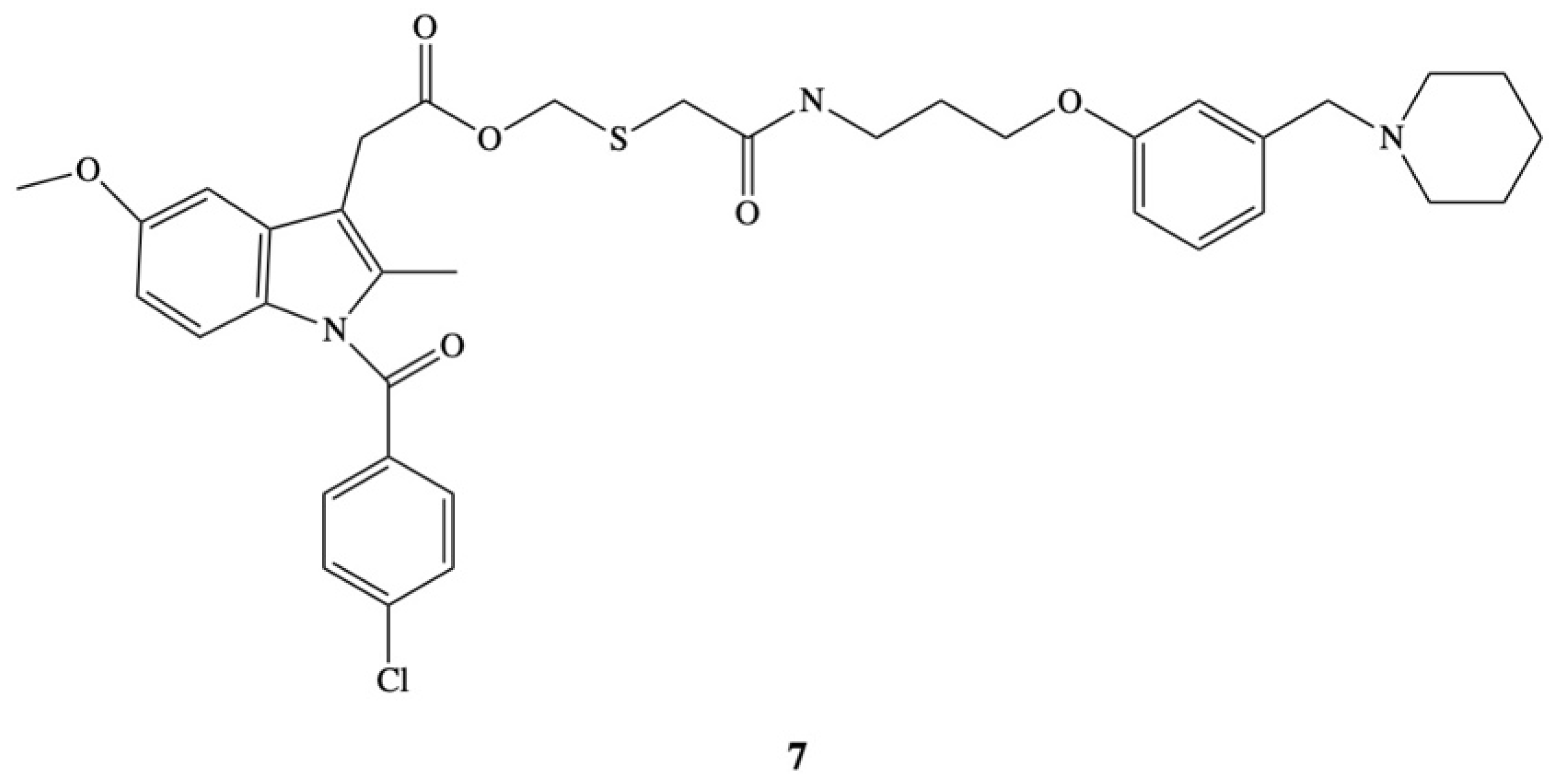
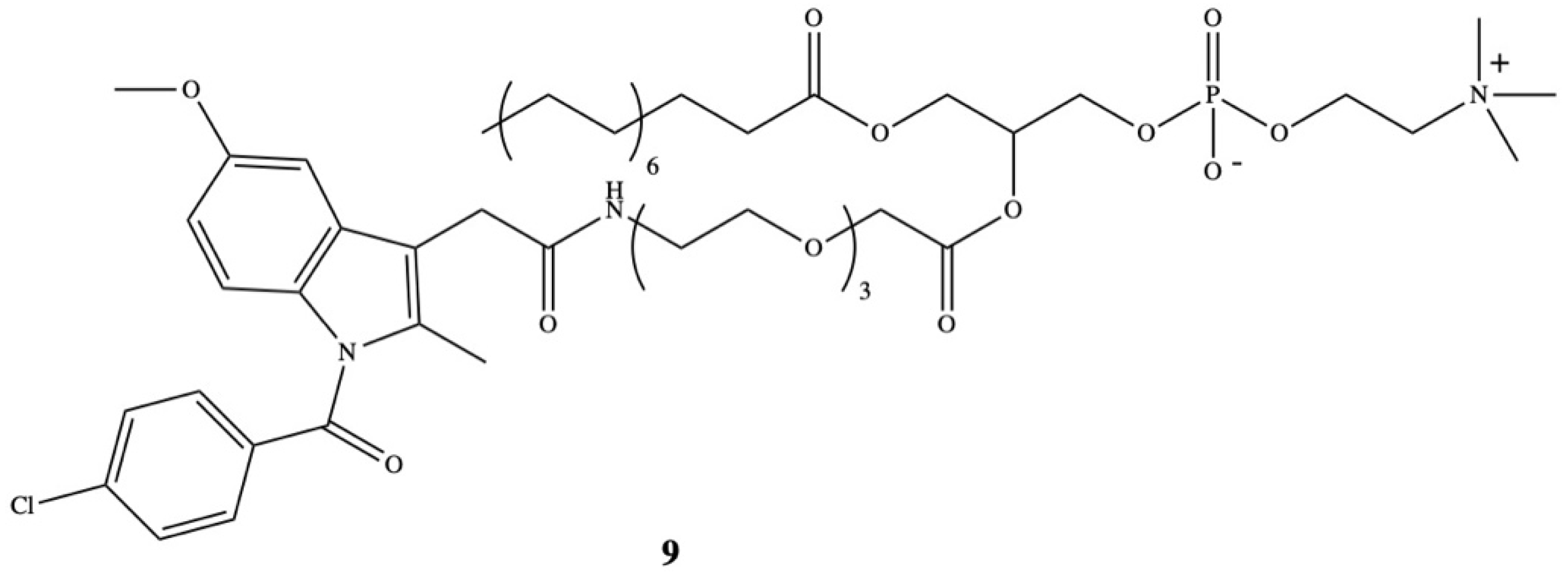
5. Bioconjugates of Indomethacin
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IND | Indomethacin |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| COX | Cyclooxygenase |
| COX-1 | Cyclooxygenase-1 |
| COX-2 | Cyclooxygenase-2 |
| GI | Gastrointestinal tract |
| DMI | O-desmethyl-indomethacin |
| DBI | O-deschlorobenzoyl-indomethacin |
| DMBI | O-desmethyl-N-deschlorobenzoyl-indomethacin |
| ACF | Aberrant Crypt Foci |
| AD | Alzheimer’s disease |
| PGs | Prostaglandins |
| CLASS | Celecoxib Long-term Arthritis Safety Study |
| VIGOR | Vioxx Gastrointestinal Outcomes Research |
| FDA | Food and Drug Administration |
| TAG | Triacylglycerol |
| PLA2 | Phospholipase A2 |
| SEDDS | Self-emulsifying Drug Delivery Systems |
| STE | Stearylamine |
References
- Dodick, D.W. Indomethacin-responsive headache syndromes. Curr. Pain Headache Rep. 2004, 8, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Green, G.A. Understanding NSAIDs: From aspirin to COX-2. Clin. Cornerstone 2001, 3, 50–59. [Google Scholar] [CrossRef]
- Lucas, S. The pharmacology of indomethacin. Headache J. Head Face Pain 2016, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.J.Y.; Leung, W.K.; Go, M.Y.Y.; To, K.F.; Cheng, A.S.L.; Ng, E.K.W.; Chan, F.K.L. Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am. J. Pathol. 2000, 157, 729–735. [Google Scholar] [CrossRef] [Green Version]
- Botting, R.; Ayoub, S.S. COX-3 and the mechanism of action of paracetamol/acetaminophen. Prostaglandins Leukot. Essent. Fat. Acids 2005, 72, 85–87. [Google Scholar] [CrossRef]
- Griswold, D.E.; Adams, J.L. Constitutive cyclooxygenase (COX-1) and inducible cyclooxygenase (COX-2): Rationale for selective inhibition and progress to date. Med. Res. Rev. 1996, 16, 181–206. [Google Scholar] [CrossRef]
- Higgs, G.A.; Flower, R.J.; Vane, J.R. A new approach to anti-inflammatory drugs. Biochem. Pharmacol. 1979, 28, 1959–1961. [Google Scholar] [CrossRef]
- Di Rosa, M.; Papadamitriou, J.M.; Willoughby, D.A. A histopathological and pharmacological analysis of the mode of action of non-steroidal anti-inflammatory drugs. J. Pathol. 1971, 105, 239–256. [Google Scholar] [CrossRef] [PubMed]
- McCall, E.; Youlten, L.F.J. The effect of indomethacin and depletion of complement on cell migration and prostaglandin levels in carragenin-induced air bleb inflammation. Br. J. Pharmacol. 1974, 52, 452. [Google Scholar]
- Ammendola, G.; Di Rosa, M.; Sorrentino, L. Leucocyte migration and lysosomal enzymes release in rat carrageenin pleurisy. Agents Actions 1975, 5, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Smith, M.J.H.; Ford-Hutchinson, A.W. Anti-inflammatory drugs, prostaglandins and leucocyte migration. Agents Actions 1976, 6, 602–606. [Google Scholar] [CrossRef]
- Yeh, K.C. Pharmacokinetic overview of indomethacin and sustained-release indomethacin. Am. J. Med. 1985, 79, 3–12. [Google Scholar] [CrossRef]
- Helleberg, L. Clinical pharmacokinetics of indomethacin. Clin. Pharmacokinet. 1981, 6, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Emori, H.W.; Champion, G.D.; Bluestone, R.; Paulus, H.E. Stimultaneous pharmacokinetics of indomethacin in serum and synovial fluid. Ann. Rheum. Dis. 1976, 32, 433–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brune, K.; Glatt, M.; Graf, P. Mechanism of action of anti-inflammatory drugs. Gen. Pharmacol. 1976, 7, 27–33. [Google Scholar] [CrossRef]
- Duggan, D.E.; Hogans, A.F.; Kwan, K.C.; McMahon, F.G. The metabolism of indomethacin in man. J. Pharmacol. Exp. Ther. 1972, 181, 563–575. [Google Scholar]
- Stoll, B.A. Indomethacin in breast cancer. Lancet 1973, 2, 384. [Google Scholar] [CrossRef]
- Waddell, W.R.; Gerner, R.E. Indomethacin and ascorbate inhibit desmoid tumors. J. Surg. Oncol. 1980, 15, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Waddell, W.R.; Cerner, R.E.; Reich, M.P. Nonsteroidal anti-inflammatory drugs and tamoxifen for desmoid tumors and carcinoma of the stomach. J. Surg. Oncol. 1983, 22, 197–211. [Google Scholar] [CrossRef]
- Charalambous, D.C.; Farmer, C.; O’Brien, P.E. Sulindac and indomethacin inhibit formation of aberrant crypt foci in the colons of dimethylhydrazine treated rats. J. Gastroenterol. Hepatol. 1996, 11, 88–92. [Google Scholar] [CrossRef]
- Brown, W.A.; Skinner, S.A.; Malcontenti-Wilson, C.; Misajon, A.; Dejong, T.; Vogiagis, D.; O’Brien, P.E. Non-steroidal anti-inflammatory drugs with different cyclooxygenase inhibitory profiles that prevent aberrant crypt foci formation but vary in acute gastrotoxicity in a rat model. J. Gastroenterol. Hepatol. 2000, 15, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Pollard, M.; Luckert, P.H. Indomethacin treatment of rats with dimethylhydrazine-induced intestinal tumors. Cancer Treat. Rep. 1980, 64, 1323–1327. [Google Scholar]
- Brown, W.A.; Skinner, S.A.; Malcontenti-Wilson, C.; Vogiagis, D.; O’Brien, P.E. Non-steroidal anti-inflammatory drugs with activity against either cyclooxygenase 1 or cyclooxygenase 2 inhibit colorectal cancer in a DMH rodent model by inducing apoptosis and inhibiting cell proliferation. Gut 2001, 48, 660–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panje, W.R. Regression of head and neck carcinoma with a prostaglandin-synthesis inhibitor. Arch. Otolaryngol. 1981, 107, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Eli, Y.; Przedecki, F.; Levin, G.; Kariv, N.; Raz, A. Comparative effects of indomethacin on cell proliferation and cell cycle progression in tumor cells grown in vitro and in vivo. Biochem. Pharmacol. 2001, 61, 565–571. [Google Scholar] [CrossRef]
- Brunelli, C.; Amici, C.; Angelini, M.; Fracassi, C.; Belardo, G.; Santoro, M.G. The non-steroidal anti-inflammatory drug indomethacin activates the eIF2α kinase PKR, causing a translational block in human colorectal cancer cells. Biochem. J. 2012, 443, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.-C.; Chan, C.-M.; Hsu, W.-L.; Chiu, S.-J.; Tsai, Y.-T.; Chou, Y.-H.; Hou, M.-F.; Wang, J.-Y.; Lee, M.-H.; Tsai, K.-L.; et al. Indomethacin inhibits cancer cell migration via attenuation of cellular calcium mobilization. Molecules 2013, 18, 6584–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, A.K.-W.; Cao, H.-H.; Cheng, C.-Y.; Kwan, H.-K.; Yu, H.; Fong, W.-F.; Yu, Z.-L. Indomethacin sensitizes TRIAL-resistant melanoma cells to TRIAL-induced apoptosis through ROS-mediated upregulation of death receptor 5 and downregulation of surviving. J. Investig. Dermatol. 2014, 134, 1397–1407. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Amstrong, C.M.; Lou, W.; Lombard, A.; Evans, C.P.; Gao, A.C. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol. Cancer Ther. 2017, 16, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Wang, B.; Fan, Q.; Xu, C.; He, Y.; Chen, Y. Chemosensitizing indomethacin-conjugated dextran-based micelles for effective delivery of paclitaxel in resistant breast cancer therapy. PLoS ONE 2017, 12, e0180037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wang, S.; Ying, X.; Wang, Y.; Geng, P.; Deng, A.; Yu, Z. Doxorubicin-loaded redox-responsive micelles based on dextran and indomethacin for resistant breast cancer. Int. J. Nanomed. 2017, 12, 6153–6168. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.; Kirby, L.C.; Hempelmman, S.R.; Berry, D.L.; McGeer, P.L.; Kaszniak, A.W.; Zalinski, J.; Cofield, M.; Mansukhani, L.; Willson, P.; et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology 1993, 43, 1609–1611. [Google Scholar] [CrossRef]
- McGerr, P.L.; McGeer, E.G. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol. Aging 2007, 28, 639–647. [Google Scholar] [CrossRef]
- Food and Drug Administration. Analysis and Recommendations for Agency Action Regarding Non-Steroidal Anti-Inflammatory Drugs and Cardiovascular Risk. 2005. Available online: http:/www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm429364.htm (accessed on 10 December 2015).
- Somasundaram, S.; Hayllar, H.; Rafi, S.; Wrigglesworth, J.M.; Macpherson, A.J.S.; Bjarnason, I. Review: The Biochemical Basis of Non-Steroidal Anti-Inflammatory Drug-Induced Damage to the Gastrointestinal Tract: A Review and a Hypothesis. Scand. J. Gastroenterol. 1995, 30, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, L.M. Where is the evidence that cyclooxygenase inhibition is the primary cause of nonsteroidal anti-inflammatory drug (NSAID)-induced gastrointestinal injury? Biochem. Pharmacol. 2001, 61, 631–637. [Google Scholar] [CrossRef]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Hunter, L.J.; Wood, D.M.; Dargan, P.I. The patterns of toxicity and management of acute nonsteroidal anti-inflammatory drug (NSAID) overdose. Open Access Emerg. Med. 2011, 3, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Gleason, C.A. Prostaglandins and the developing kidney. Semin. Perinatol. 1987, 11, 12–21. [Google Scholar]
- Olesen, E.T.B.; Fenton, R.A. Is There a Role for PGE2 in Urinary Concentration? J. Am. Soc. Nephrol. 2012, 24, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Bolger, P.M.; Eisner, G.M.; Shea, P.T.; Ramwell, P.W.; Slotkoff, L.M. Effects of PGD 2 on canine renal function. Nature 1977, 267, 628–630. [Google Scholar] [CrossRef]
- Ejaz, P.; Bhojani, K.; Joshi, V.R. NSAIDs and kidney. JAPI 2004, 52, 371. [Google Scholar]
- Calvo-Alen, J.; De, M.C.; Rodriguez-Valverde, V.; Escallada, R.; Florez, J.; Arias, M. Subclinical renal toxicity in rheumatic patients receiving longterm treatment with nonsteroidal antiinflammatory drugs. J. Rheumatol. 1994, 21, 1742–1747. [Google Scholar] [PubMed]
- Pathan, E.; Gaitonde, S.; Rajadhyaksha, S.; Sule, A.; Mittal, G.; Joshi, V.R. A longitudinal study of serum creatinine levels in patients of rheumatoid arthritis on long term NSAID therapy. J. Assoc. Phys. India 2003, 51, 1045–1051. [Google Scholar]
- Nuutinen, L.S.; Laitinen, J.O.; Salomäki, T.E. A Risk-Benefit Appraisal of Injectable NSAIDs in the Management of Postoperative Pain. Drug Saf. 1993, 9, 380–393. [Google Scholar] [CrossRef]
- Lane, N.E. Pain management in osteoarthritis: The role of COX-2 inhibitors. J. Rheumatol. Suppl. 1997, 49, 20–24. [Google Scholar] [PubMed]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Dar, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal toxicity with celecoxib vs. nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 2001, 286, 954–959. [Google Scholar] [CrossRef]
- Praveen Rao, P.N.; Knaus, E.E. Evolution of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): Cyclooxygenase (COX) Inhibition and Beyond. J. Pharm. Pharm. Sci. 2008, 11, 81s–110s. [Google Scholar]
- Wright, J.M. The double-edged sword of COX-2 selective NSAIDs. CMAJ 2002, 167, 1131–1137. [Google Scholar] [PubMed]
- Zhou, Y.; Hancock, J.F.; Lichtenberger, L.M. The nonsteroidal anti-inflammatory drug indomethacin induces heterogeneity in lipid membranes: Potential implication for its diverse biological action. PLoS ONE 2010, 5, e8811. [Google Scholar] [CrossRef]
- Fearon, A.; Stokes, G.Y. Thermodynamics of indomethacin adsorption to phospholipid membranes. J. Phys. Chem. B 2017, 121, 10508–10518. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Wang, Z.M.; Romero, J.J.; Ulloa, C.; Perez, J.C.; Giraud, M.N.; Barreto, J.C. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat. Med. 1995, 1, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Dial, E.J.; Dawson, P.A.; Lichtenberger, L.M. In vitro evidence that phosphatidylcholine protects against indomethacin/bile acid-induced injury to cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G217–G222. [Google Scholar] [CrossRef] [Green Version]
- Lichtenberger, L.M.; Phan, T.; Fang, D.; Dial, E.J. Chemoprevention with phosphatidylcholine non-steroidal anti-inflammatory drugs in vivo and in vitro. Oncol. Lett. 2018, 15, 6688–6694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.J.; Phan, T.M.; Dial, E.J.; Graham, D.; Lichtenberger, L.M. In vitro and in vivo protection against indomethacin-induced small intestinal injury by proton pump inhibitors, acid pump antagonists, or indomethacin-phosphatidylcholine. Digestion 2012, 86, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, R.A.; Tramer, M.R.; Carroll, D.; Wiffen, P.J.; McQuay, H.J. Quantitative systematic review of topically applied non-steroidal anti-inflammatory drugs. BMJ 1998, 316, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Cordero, J.A.; Alarcon, L.; Escribano, E.; Obach, R.; Domenech, J. A comparative study of the transdermal penetration of a series of nonsteroidal anti-inflammatory drugs. J. Pharm. Sci. 1997, 86, 503–508. [Google Scholar] [CrossRef]
- Goosen, C.; Du Plessis, J.; Muller, D.G.; Janse van Rensburg, L.F. Correlation between physicochemical characteristics, pharmacokinetic properties and transdermal absorption of NSAID’s. Int. J. Pharm. 1998, 163, 203–209. [Google Scholar] [CrossRef]
- Sakdiset, P.; Amnuaikit, T.; Pichayakorn, W.; Pinsuwan, S. Formulation development of ethosomes containing indomethacin for transdermal delivery. J. Drug Deliv. Sci. Technol. 2019, 52, 760–768. [Google Scholar] [CrossRef]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ku, Y.S. Enhanced absorption of indomethacin after oral or rectal administration of a self-emulsifying system containing indomethacin to rats. Int. J. Pharm. 2000, 194, 81–89. [Google Scholar] [CrossRef]
- Manosroi, A.; Wongtrakul, P.; Manosroi, J.; Sakai, H.; Sugawara, F.; Yuasa, M.; Abe, M. Characterization of vesicles prepared with various non-ionic surfactants mixed with cholesterol. Colloids Surf. B Biointerfaces 2003, 30, 129–138. [Google Scholar] [CrossRef]
- Cereda, S.; De Araujo, D.R.; De Paula, E. Liposomal prilocaine: Preparation, characterization, and in vivo evaluation. J. Pharm. Pharm. Sci. 2004, 7, 235–240. [Google Scholar] [PubMed]
- Soehngen, E.C.; Godin-Ostro, E.; Fielder, F.G.; Ginsberg, R.S.; Slusher, M.A.; Weiner, A.L. Encapsulation of indomethacin in liposomes provides protection against both gastric and intestinal ulceration when orally administered to rats. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1988, 31, 414–422. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, J.; Notari, R.E. Drug stability in liposomal suspensions: Hydrolysis of indomethacin, cyclocytidine and p-nitrophenyl acetate. J. Pharm. Sci. 1982, 71, 1394–1398. [Google Scholar] [CrossRef]
- Hernandez, J.; Estelrich, J.; Montero, M.T.; Valls, O. The action of indomethacin on neutral and positively charged monolayers. Colloid Polym. Sci. 1989, 267, 622–626. [Google Scholar] [CrossRef]
- Srinath, P.; Vyas, S.P.; Diwan, P.V. Effect of lipid composition of liposomes on biodisposition of indomethacin in arthritic rats. Pharm. Pharmacol. Commun. 1999, 5, 339–344. [Google Scholar] [CrossRef]
- Srinath, P.; Chary, M.G.; Vyas, S.P.; Diwan, P.V. Long-circulating liposomes of indomethacin in arthritic rats—A biodisposition study. Pharm. Acta Helv. 2000, 74, 399–404. [Google Scholar] [CrossRef]
- Srinath, P.; Vyas, S.P.; Diwan, P.V. Preparation and pharmacodynamic evaluation of liposomes of indomethacin. Drug Dev. Ind. Pharm. 2000, 26, 313–321. [Google Scholar] [CrossRef]
- Sugihura, H.; Yamamoto, H.; Kawashima, Y.; Takeuchi, H. Effectiveness of submicronized chitosan-coated liposomes in oral absorption of indomethacin. J. Liposome Res. 2012, 22, 285–294. [Google Scholar] [CrossRef]
- Refuerzo, J.S.; Alexander, J.F.; Leonard, F.; Leon, M.; Longo, M.; Godin, B. Liposomes: A nanoscale drug carrying system to prevent indomethacin passage to the fetus in a pregnant mouse model. Am. J. Obstet. Gynecol. 2015, 212, 508.e1. [Google Scholar] [CrossRef]
- Refuerzo, J.S.; Leonard, F.; Bulayeva, N.; Gorenstein, D.; Chiossi, G.; Ontiveros, A.; Longo, M.; Godin, B. Uterus-targeted liposomes for preterm labor management: Studies in pregnant mice. Sci. Rep. 2016, 6, 34710. [Google Scholar] [CrossRef] [Green Version]
- Nihala, N.; Mathan, S.; Rajalekshmi, V.R.; Bineesha, K.B. Development of formulation and in vitro evaluation of sterically stabilized (stealth) liposomes containing selected anti-arthritic drug. J. Pharm. Sci. Res. 2019, 11, 3526–3535. [Google Scholar]
- Yáñez, J.A.; Wang, S.W.J.; Knemeyer, I.W.; Wirth, M.A.; Alton, K.B. Intestinal lymphatic transport for drug delivery. Adv. Drug Deliv. Rev. 2015, 63, 923–942. [Google Scholar] [CrossRef]
- Pedersen, P.J.; Christensen, M.S.; Ruysschaert, T.; Linderoth, L.; Andresen, T.L.; Melander, F.; Mouritsen, O.G.; Madsen, R.; Clausen, M.H. Synthesis and Biophysical Characterization of Chlorambucil Anticancer Ether Lipid Prodrugs. J. Med. Chem. 2009, 52, 3408–3415. [Google Scholar] [CrossRef]
- Liu, H.; Bolleddula, J.; Nichols, A.; Tang, L.; Zhao, Z.; Prakash, C. Metabolism of bioconjugate therapeutics: Why, when, and how? Drug Metab. Rev. 2020, 52, 66–124. [Google Scholar] [CrossRef]
- Paris, G.Y.; Garmaise, D.L.; Cimon, D.G.; Swett, L.; Carter, G.W.; Young, P. Glycerides as prodrugs. 3. Synthesis and anti-inflammatory activity of [1-(p-chlorobenzoyl)-5-methoxy-2-methylindole-3-acetyl]glycerides (indomethacin glycerides). J. Med. Chem. 1980, 23, 9–13. [Google Scholar] [CrossRef]
- Paris, G.Y.; Cimon, D.G. Glycerides as prodrugs. 4. Synthesis and anti-inflammatory activity of 1,3-dialkananoyl-2-arylalkanoylglycerides. Eur. J. Med. Chem. 1982, 17, 193–195. [Google Scholar]
- Paris, G.Y.; Garmaise, D.L.; Cimon, D.G.; Swett, L.; Carter, G.W.; Young, P. Glycerides as prodrugs. 1. Synthesis and anti-inflammatory activity of 1,3-bis(alkanoyl)-2-(O-acetylsalicyloyl)glycerides (aspirin triglycerides). J. Med. Chem. 1979, 22, 683–687. [Google Scholar] [CrossRef]
- Carter, G.W.; Young, P.R.; Swett, L.R.; Paris, G.Y. Pharmacological studies in the rat with [2-(1,3-didecanoyloxy)-propyl]2-acetyloxybenzoate (A-45474): An aspirin pro-drug with negligible gastric irritation. Agents Actions 1980, 10, 240–245. [Google Scholar] [CrossRef]
- Ueda, I.; Ishii, K.; Arai, H.; Ikeda, S.; Hitomi, Y.; Hatanaka, M. Design, synthesis and anti-inflammatory activity of a new indomethacin ester. 2-[N-[3-{3-(Piperodonomethyl)phenoxy}propyl]-carbamoylmethylthio]ethyl 1-(p-chlorobenzoyl)-5-methoxy-2-methylindole-3-acetate. Chem. Pharm. Bull. 1991, 39, 679–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvir, E.; Friedman, E.; Lee, J.Y.; Koh, J.Y.; Younis, F.; Raz, S.; Shapiro, I.; Hoffman, A.; Dahan, A.; Rosenberg, G.; et al. A Novel Phospholipid Derivative of Indomethacin, DP-155 [Mixture of 1-Steroyl and 1-Palmitoyl-2-{6-[1-(p-chlorobenzoyl)-5-methoxy-2-methyl-3-indolyl acetamido]butanoyl}-sn-glycero-3-phosophatidyl Choline], Shows Superior Safety and Similar Efficacy in Reducing Brain Amyloid β in an Alzheimer’s Disease Model. J. Pharmacol. Exp. Ther. 2006, 318, 1248–1256. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, J.; Kelly, P.A.; Grome, J.J.; Pickard, J.D. Local cerebral circulatory and metabolic effects of indomethacin. Am. J. Physiol. Heart Circ. Physiol. 1982, 243, H416–H423. [Google Scholar] [CrossRef]
- Markus, H.S.; Vallance, P.; Brown, M.M. Differential effect of three cyclooxygenase inhibitors on human cerebral blood flow velocity and carbon dioxide reactivity. Stroke 1994, 25, 1760–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahan, A.; Hoffman, A. Mode of Administration-Dependent Brain Uptake of Indomethacin: Sustained Systemic Input Increases Brain Influx. Drug Metab. Dispos. 2006, 35, 321–324. [Google Scholar] [CrossRef]
- Dahan, A.; Duvdevani, R.; Dvir, E.; Elmann, A.; Hoffman, A. A novel mechanism for oral controlled release of drugs by continuous degradation of a phospholipid prodrug along the intestine: In-vivo and in-vitro evaluation of an indomethacin–lecithin conjugate. J. Control. Release 2007, 119, 86–93. [Google Scholar] [CrossRef]
- Dvir, E.; Elman, A.; Simmons, D.; Shapiro, I.; Duvdevani, R.; Dahan, A.; Hoffman, A.; Friedman, J.E. DP-155, a Lecithin Derivative of Indomethacin, is a Novel Nonsteroidal Antiinflammatory Drug for Analgesia and Alzheimer’s Disease Therapy. CNS Drug Rev. 2007, 13, 260–277. [Google Scholar] [CrossRef]
- Graff, J.R.; Konicek, B.W.; Deddens, J.A.; Chedid, M.; Hurst, B.M.; Colligan, B.; Neubaer, B.L.; Carter, H.W.; Carter, J.H. Expression of IIa secretory phospholipase A2 increases with prostate tumor grade. Clin. Cancer Res. 2001, 7, 3857–3861. [Google Scholar]
- Menschikowski, M.; Hagelgans, A.; Gussakovski, E.; Kostka, H.; Paley, E.L.; Siegert, G. Differential expression of phospholipase A2 in normal and malignant prostate cell lines: Regulation by cytokines, cell signaling pathways and epigenic mechanism. Neoplasia 2008, 10, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosseto, R.; Hajdu, J. Synthesis of oligo(ethylene glycol) substituted phosphatidylcholines: Secretory PLA2-targeted precursors of NSAID prodrugs. Chem. Phys. Lipids 2010, 163, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Model | Active Dose | In Vitro/In Vivo Outcomes | Reference |
|---|---|---|---|
| Patients with desmoid tumors | 100 mg/day in combination with ascorbic acid | Inhibition of a desmoid tumor/ postulate that lowering of cAMP inhibits cell growth | [18] |
| Patients with desmoid tumors | Alone or in combination with 5-fluorouracil and cyclophosphamide | Inhibition of suppressor T-cells | [19] |
| Dimethylhydrazine (DMH)-induced rats | 2 mg/kg per day | Inhibition of formation of aberrant crypt foci (ACF)/significant inhibition of growth and development of tumors | [20] |
| Dimethylhydrazine (DMH)-induced rats | 2 mg/kg per day | Reduction of 42% of number of ACF | [21] |
| Dimethylhydrazine (DMH)-induced rats (colorectal tumors) | 2 mg/kg per day | Reduction of tumor number (83.5%), reduction of tumor volume (95%), increase of rate of apoptosis and reduction of proliferation in the S phase | [23] |
| Patients with head and neck cancer (stage III and IV) | 75–100 mg/day | Tumor regression, increase of survival | [24] |
| Lung carcinoma cells mouse | 10–20 μM 2 mg/kg per day | Reduction of cells at the S and G2/M phases and increase of cells at G1 phase inhibition of COX activity and effective in delaying the growth of both the primary tumor inoculate and of lung metastatic nodules | [25] |
| Human colon carcinoma (HT29, HCT116, Caco-2), lung adenocarcinoma (A549), cervical adenocarcinoma (HeLa) cells | 400–1000 μM | Selective activation of dsRNA (double-stranded RNA)-dependent protein kinase PKR in a cyclooxygenase-independent manner, rapid phosphorylation of eIF2α and inhibition protein synthesis in carcinoma, induction of apoptosis | [26] |
| Human epidermoid carcinoma (A431) | 1–10 μM | Inhibition of cancer cell migration by influencing calcium mobilization and focal complex formation | [27] |
| Melanoma cells (A375) | 1–300 μM | Promotion of TRIAL-induced cell death and apoptosis, induction of cell surface expression of death receptor 5 (DR5) | [28] |
| Prostate cancer cells | 2.5–10 μM | Inhibition of activity of enzyme in the steroidogenesis pathway AKR1C3 through binding with its active site, strong selectivity for AKR1C3 at 8.2 μM over AKR1C1 and AKR1C2 (over 100 μM), inhibition of the levels of intracrine androgens in C4-2B MDVR cells and CWR22Rv1 cells and suppression of prostate cancer tumor growth | [29] |
| Liposomal Type | Physiochemical Characteristics | Study Model | In Vivo Outcomes | Reference |
|---|---|---|---|---|
| IND encapsulated into egg PC (EPC) monophasic vesicles (MPV) and into stable plurilamellar vesicles (SPLV) | Spherical structures, size range of 0.5 μm | Male Wistar rats | EPCMPV containing IND (4 mg/kg) reduced gastric and intestinal ulceration, anti-inflammatory effect with a dosage ranging between 0.5 and 4 mg/kg | [66] |
| IND encapsulated in liposomes prepared with usage of various phospholipids (PC, PE, PG), stearylamine (SA) and cholesterol (CH) | The highest encapsulation efficiency 32% for liposome composition PC:CH:SA (1:0.5:0.1 molar ratio), | Male Wistar rats | Cmax in the liver delayed from 1 h for free drug to 4 h for encapsulated form, localization in the liver was greatest for liposomes PC:CH:PG (1:0.5:0.2 molar ratio), this composition is the optimum for targeting arthritic joints | [69] |
| Long-circulating liposomes (S-LI) | Encapsulation efficiency 52–55%, liposome composition PC:CH:PE-PEG (1:0.5:0.16) | Male Wistar rats | Better pharmacokinetic profile (AUC0−t 1454.62 ± 92.85 μg/mL/h, elimination half-life 25.42 ± 4.32 h and clearance 0.82 ± 0.15 mL/h, MRT 36.36 6.25 h) than free IND (AUC0−t 490.95 ± 31.28 μg/mL/h, elimination half-life 10.28 ± 0.25 h and clearance 4.20 0.33 mL/h, MRT 13.27 0.49 h) Increased anti-inflammatory activity, less ulcer index | [70] |
| Chitosan-coated liposomes | Liposome composition DSPC:DCP:CH (8:2:1) coating with chitosan, Liposomal dispersion, Size 270–310 nm | Rats | Prolonged intestinal transit, delayed drug release profile | [72] |
| Multilamellar liposomes | Size 159.8 nm Polydispersity index < 0.069 Encapsulation efficiency 93% | Rats | Reduction of the drug levels within the fetus by 7.6-fold yet maintained its pharmacologic effects | [73] |
| IND loaded in liposomes with oxytocin receptor antagonist (LIP-IND-ORA) | Size 154.2 nm Zeta potential −21.2 mV Encapsulation efficiency 93% | Rats | Uterine to fetus IND concentration ratio was 4-fold higher for liposomes than for free drug, encapsulation of IND does not alter the pharmacological activity of drug | [74] |
| Sterically stabilized liposomes | Zeta potential −35.3 mV Encapsulation efficiency 64.04–79.54% | Rats | Increased in vitro drug release in comparison to the conventional liposomal formulation, better in vivo circulation time and enhanced mean percentage edema decrease for stealth liposomes in comparison with conventional liposomes and drug, higher stability (3 months) | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gliszczyńska, A.; Nowaczyk, M. Lipid Formulations and Bioconjugation Strategies for Indomethacin Therapeutic Advances. Molecules 2021, 26, 1576. https://doi.org/10.3390/molecules26061576
Gliszczyńska A, Nowaczyk M. Lipid Formulations and Bioconjugation Strategies for Indomethacin Therapeutic Advances. Molecules. 2021; 26(6):1576. https://doi.org/10.3390/molecules26061576
Chicago/Turabian StyleGliszczyńska, Anna, and Marta Nowaczyk. 2021. "Lipid Formulations and Bioconjugation Strategies for Indomethacin Therapeutic Advances" Molecules 26, no. 6: 1576. https://doi.org/10.3390/molecules26061576
APA StyleGliszczyńska, A., & Nowaczyk, M. (2021). Lipid Formulations and Bioconjugation Strategies for Indomethacin Therapeutic Advances. Molecules, 26(6), 1576. https://doi.org/10.3390/molecules26061576