
The Fascinating Flexibility and Coordination Modes of a Pentamethylene Connected Macrocyclic CNC Pincer Ligand


Abstract
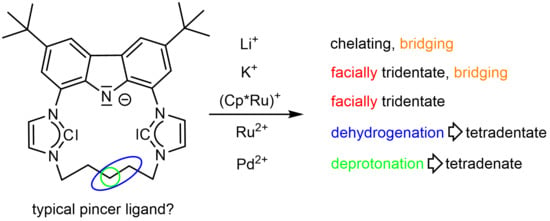
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Alkali Metal bimcaC5 Complexes
2.2. Ruthenium(II) bimcaC5 Complexes
2.2.1. Facial Coordination of the bimcaC5 Ligand
2.2.2. Complex Formation under Dehydrogenation of bimcaC5
2.3. A macrocyclic Palladium(II) bimcaC5 Complex by C-H Activation
3. Materials and Methods
3.1. Preparation of (HbimcaC5)·2HPF6 (1b)
3.2. General Procedure for the Generation of Alkali Metal bimcaC5 Complexes
3.3. Synthesis of the Ru(II) Sandwich Complex 4 (Ru(bimcaC5)Cp*)
3.4. Synthesis of the Macrocyclic Ru(II)(bimcaC5) Complex 5
3.5. Preparation of the Macrocyclic Pd(II) Complex 6
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hood, T.M.; Gyton, M.R.; Chaplin, A.B. Synthesis and rhodium complexes of macrocyclic PNP and PONOP pincer ligands. Dalton Trans. 2020, 49, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Leforestier, B.; Gyton, M.R.; Chaplin, A.B. Synthesis and group 9 complexes of macrocyclic PCP and POCOP pincer ligands. Dalton Trans. 2020, 49, 2087–2101. [Google Scholar] [CrossRef]
- Andrew, R.E.; Chaplin, A.B. Synthesis, structure and dynamics of NHC-based palladium macrocycles. Dalton Trans. 2014, 43, 1413–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, R.E.; Chaplin, A.B. Synthesis and reactivity of NHC-based rhodium macrocycles. Inorg. Chem. 2015, 54, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.E.; Storey, C.M.; Chaplin, A.B. Well-defined coinage metal transfer agents for the synthesis of NHC-based nickel, rhodium and palladium macrocycles. Dalton Trans. 2016, 45, 8937–8944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, C.M.; Gyton, M.R.; Andrew, R.E.; Chaplin, A.B. Terminal Alkyne Coupling Reactions through a Ring: Mechanistic Insights and Regiochemical Switching. Angew. Chem. Int. Ed. 2018, 57, 12003–12006. [Google Scholar] [CrossRef]
- Leforestier, B.; Gyton, M.R.; Chaplin, A.B. Oxidative Addition of a Mechanically Entrapped C(sp)-C(sp) Bond to a Rhodium(I) Pincer Complex. Angew. Chem. 2020, 59, 23500–23504. [Google Scholar] [CrossRef]
- Storey, C.M.; Gyton, M.R.; Andrew, R.E.; Chaplin, A.B. Terminal Alkyne Coupling Reactions Through a Ring: Effect of Ring Size on Rate and Regioselectivity. Chem. Eur. J. 2020, 26, 14715–14723. [Google Scholar] [CrossRef]
- Biffis, A.; Cipani, M.; Bressan, E.; Tubaro, C.; Graiff, C.; Venzo, A. Group 10 Metal Complexes with Chelating Macrocyclic Dicarbene Ligands Bearing a 2,6-Lutidinyl Bridge: Synthesis, Reactivity, and Catalytic Activity. Organometallics 2014, 33, 2182–2188. [Google Scholar] [CrossRef]
- Alcalde, E.; Ramos, S.; Perez-Garcia, L. Anion Template-Directed Synthesis of Dicationic [14]Imidazoliophanes. Org. Lett. 1999, 1, 1035–1038. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, G.; Jiang, Z.-L.; You, J.-S.; Zhou, Z.-Y.; Yuan, D.-Q.; Xie, R.-G. Synthesis and selective anion recognition of imidazolium cyclophanes. Tetrahedron 2002, 58, 8993–8999. [Google Scholar] [CrossRef]
- Radloff, C.; Gong, H.-Y.; Schulte to Brinke, C.; Pape, T.; Lynch, V.M.; Sessler, J.L.; Hahn, F.E. Metal-Dependent Coordination Modes Displayed by Macrocyclic Polycarbene Ligands. Chem. Eur. J. 2010, 16, 13077–13081. [Google Scholar] [CrossRef] [PubMed]
- Jürgens, E.; Buys, K.N.; Schmidt, A.-T.; Furfari, S.K.; Cole, M.L.; Moser, M.; Rominger, F.; Kunz, D. Optimised synthesis of monoanionic bis(NHC)-pincer ligand precursors and their Li-complexes. New J. Chem. 2016, 40, 9160–9169. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Yang, C.-F.; Steren, C.A.; Fei, F.; Chen, X.-T.; Xue, Z.-L. Synthesis and characterization of Ag(I) and Au(I) complexes with macrocyclic hybrid amine N-heterocyclic carbene ligands. New J. Chem. 2018, 42, 4700–4713. [Google Scholar] [CrossRef]
- Moser, M.; Wucher, B.; Kunz, D.; Rominger, F. 1,8-Bis(imidazolin-2-yliden-1-yl)carbazolide (bimca): A New CNC Pincer-Type Ligand with Strong Electron-Donating Properties. Facile Oxidative Addition of Methyl Iodide to Rh(bimca)(CO). Organometallics 2007, 26, 1024–1030. [Google Scholar] [CrossRef]
- Evans, K.J.; Campbell, C.L.; Haddow, M.F.; Luz, C.; Morton, P.A.; Mansell, S.M. Lithium Complexes with Bridging and Terminal NHC Ligands: The Decisive Influence of an Anionic Tether. Eur. J. Inorg. Chem. 2019, 2019, 4894–4901. [Google Scholar] [CrossRef]
- Simler, T.; Karmazin, L.; Bailly, C.; Braunstein, P.; Danopoulos, A.A. Potassium and Lithium Complexes with Monodeprotonated, Dearomatized PNP and PNC NHC Pincer-Type Ligands. Organometallics 2016, 35, 903–912. [Google Scholar] [CrossRef]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018, 118, 9678–9842. [Google Scholar] [CrossRef]
- Tapu, D.; Dixon, D.A.; Roe, C. 13C NMR spectroscopy of “Arduengo-type” carbenes and their derivatives. Chem. Rev. 2009, 109, 3385–3407. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, A.J., III; Gamper, S.F.; Tamm, M.; Calabrese, J.C.; Davidson, F.; Craig, H.A. A Bis(carbene)-Proton Complex: Structure of a C-H-C Hydrogen Bond. J. Am. Chem. Soc. 1995, 117, 572–573. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Runte, O.; Artus, G. Synthesis and structure of an ionic beryllium-“carbene” complex. J. Organomet. Chem. 1995, 501, C1–C4. [Google Scholar] [CrossRef]
- Simler, T.; Danopoulos, A.A.; Braunstein, P. N-Heterocyclic carbene-phosphino-picolines as precursors of anionic ‘pincer’ ligands with dearomatised pyridine backbones; transmetallation from potassium to chromium. Chem. Commun. 2015, 51, 10699–10702. [Google Scholar] [CrossRef] [PubMed]
- Seyboldt, A.; Wucher, B.; Hohnstein, S.; Eichele, K.; Rominger, F.; Törnroos, K.W.; Kunz, D. Evidence for the Formation of Anionic Zerovalent Group 10 Complexes as Highly Reactive Intermediates. Organometallics 2015, 34, 2717–2725. [Google Scholar] [CrossRef]
- Jürgens, E.; Kunz, D. A Rigid CNC Pincer Ligand Acting as a Tripodal Cp Analogue. Eur. J. Inorg. Chem. 2017, 2017, 233–236. [Google Scholar] [CrossRef]
- Brookhart, M.; Green, M.L.H.; Parkin, G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. USA 2007, 104, 6908–6914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, W.; Ito, J.; Yamashita, M. CNC-pincer iron complexes containing a bis(N-heterocyclic carbene)Amido ligand: Synthesis and application to catalytic hydrogenation of alkenes. J. Organomet. Chem. 2020, 923, 121436. [Google Scholar] [CrossRef]
- Gusev, D.G.; Lough, A.J. Double C−H Activation on Osmium and Ruthenium Centers: Carbene vs Olefin Products. Organometallics 2002, 21, 2601–2603. [Google Scholar] [CrossRef]
- Chatt, J.; Watson, H.R. 491. Complexes of zerovalent transition metals with the ditertiary phosphine, Me2P·CH2 ·CH2 ·PMe2. J. Chem. Soc. 1962, 2545–2549. [Google Scholar] [CrossRef]
- Chatt, J.; Davidson, J.M. 154. The tautomerism of arene and ditertiary phosphine complexes of ruthenium(0), and the preparation of new types of hydrido-complexes of ruthenium(II). J. Chem. Soc. 1965, 843–855. [Google Scholar] [CrossRef]
- Kakiuchi, F.; Murai, S. Catalytic C-H/Olefin Coupling. Acc. Chem. Res. 2002, 35, 826–834. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalyzed C-H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L. Carboxylate-assisted transition-metal-catalyzed C-H bond functionalizations: Mechanism and scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H. Organometallic alkane CH activation. J. Organomet. Chem. 2004, 689, 4083–4091. [Google Scholar] [CrossRef]
- Zhou, X.; Malakar, S.; Zhou, T.; Murugesan, S.; Huang, C.; Emge, T.J.; Krogh-Jespersen, K.; Goldman, A.S. Catalytic Alkane Transfer Dehydrogenation by PSP-Pincer-Ligated Ruthenium. Deactivation of an Extremely Reactive Fragment by Formation of Allyl Hydride Complexes. ACS Catal. 2019, 9, 4072–4083. [Google Scholar] [CrossRef]
- Gruver, B.C.; Adams, J.J.; Warner, S.J.; Arulsamy, N.; Roddick, D.M. Acceptor Pincer Chemistry of Ruthenium: Catalytic Alkane Dehydrogenation by (CF3PCP)Ru(cod)(H). Organometallics 2011, 30, 5133–5140. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, W.; Fang, H.; Hu, A.; Huang, Z. Catalytic alkane dehydrogenations. Sci. Bull. 2015, 60, 1316–1331. [Google Scholar] [CrossRef] [Green Version]
- Chatt, J.; Hart, F.A.; Watson, H.R. 490. Complex compounds of ditertiary phosphines and arsines with nickel(0) and palladium(0). J. Chem. Soc. 1962, 2537–2545. [Google Scholar] [CrossRef]
- Al-Salem, N.A.; Empsall, H.D.; Markham, R.; Shaw, B.L.; Weeks, B. Formation of large chelate rings and cyclometallated products from diphosphines of type But2P(CH2)n PBut2(n = 5–8) and Ph2P(CH2)5PPh2 with palladium and platinum chlorides: Factors affecting the stability and conformation of large chelate rings. J. Chem. Soc. Dalton Trans. 1979, 1972–1982. [Google Scholar] [CrossRef]
- Rousseaux, S.; Gorelsky, S.I.; Chung, B.K.W.; Fagnou, K. Investigation of the mechanism of C(sp3)-H bond cleavage in Pd(0)-catalyzed intramolecular alkane arylation adjacent to amides and sulfonamides. J. Am. Chem. Soc. 2010, 132, 10692–10705. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Recent developments in SHELX. Acta Crystallogr. A Found. Crystallogr. 2013, 69, s74. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Platon Squeeze: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Steffen, C.; Thomas, K.; Huniar, U.; Hellweg, A.; Rubner, O.; Schroer, A. TmoleX—A graphical user interface for TURBOMOLE. J. Comput. Chem. 2010, 31, 2967–2970. [Google Scholar] [CrossRef]
- University of Karlsruhe and Forschungszentrum Karlsruhe GmbH. TURBOMOLE. 2011. Available online: https://www.turbomole.org/ (accessed on 21 January 2021).
- Treutler, O.; Ahlrichs, R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- von Arnim, M.; Ahlrichs, R. Performance of parallel TURBOMOLE for density functional calculations. J. Comput. Chem. 1998, 19, 1746–1757. [Google Scholar] [CrossRef]
- van Wüllen, C. Shared-memory parallelization of the TURBOMOLE programs AOFORCE, ESCF, and EGRAD: How to quickly parallelize legacy code. J. Comput. Chem. 2011, 32, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Deglmann, P.; Furche, F.; Ahlrichs, R. An efficient implementation of second analytical derivatives for density functional methods. Chem. Phys. Lett. 2002, 362, 511–518. [Google Scholar] [CrossRef]
- Deglmann, P.; Furche, F. Efficient characterization of stationary points on potential energy surfaces. J. Chem. Phys. 2002, 117, 9535–9538. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Armbruster, M.K.; Weigend, F.; van Wüllen, C.; Klopper, W. Self-consistent treatment of spin-orbit interactions with efficient Hartree-Fock and density functional methods. Phys. Chem. Chem. Phys. 2008, 10, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Middendorf, N.; Weigend, F.; Reiher, M. An efficient implementation of two-component relativistic exact-decoupling methods for large molecules. J. Chem. Phys. 2013, 138, 184105. [Google Scholar] [CrossRef] [Green Version]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials (Chem. Phys. Letters 240 (1995) 283–290). Chem. Phys. Lett. 1995, 242, 652–660. [Google Scholar] [CrossRef]
- Deglmann, P.; May, K.; Furche, F.; Ahlrichs, R. Nuclear second analytical derivative calculations using auxiliary basis set expansions. Chem. Phys. Lett. 2004, 384, 103–107. [Google Scholar] [CrossRef]
- Weigend, F. A fully direct RI-HF algorithm: Implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285–4291. [Google Scholar] [CrossRef]
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. Theory Comput. Modeling (Theor. Chim. Acta) 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordan, R.; Kunz, D. The Fascinating Flexibility and Coordination Modes of a Pentamethylene Connected Macrocyclic CNC Pincer Ligand. Molecules 2021, 26, 1669. https://doi.org/10.3390/molecules26061669
Jordan R, Kunz D. The Fascinating Flexibility and Coordination Modes of a Pentamethylene Connected Macrocyclic CNC Pincer Ligand. Molecules. 2021; 26(6):1669. https://doi.org/10.3390/molecules26061669
Chicago/Turabian StyleJordan, Ronja, and Doris Kunz. 2021. "The Fascinating Flexibility and Coordination Modes of a Pentamethylene Connected Macrocyclic CNC Pincer Ligand" Molecules 26, no. 6: 1669. https://doi.org/10.3390/molecules26061669
APA StyleJordan, R., & Kunz, D. (2021). The Fascinating Flexibility and Coordination Modes of a Pentamethylene Connected Macrocyclic CNC Pincer Ligand. Molecules, 26(6), 1669. https://doi.org/10.3390/molecules26061669