
(Trifluoromethoxy)Phenylboronic Acids: Structures, Properties, and Antibacterial Activity

 , and
, and
Abstract
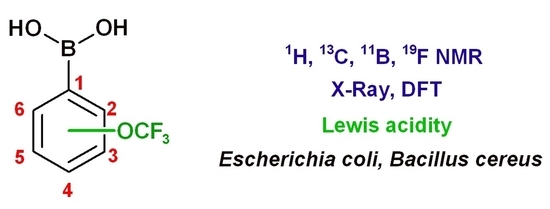
:
1. Introduction
2. Results and Discussion
2.1. Acidity and Stability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | X | ortho | meta | para | Ref. |
| OCF3 | 9.51 ± 0.04 9.49 ± 0.08 | 7.79 ± 0.02 7.96 ± 0.07 | 8.11 ± 0.04 8.03 ± 0.07 | this work | |
| OCH3 | 9.31 ± 0.02 [31] | 8.46 [32] | No data | [31,32] | |
| CF3 | 9.45 ± 0.01 9.58 ± 0.16 | 7.88 ± 0.01 7.85 ± 0.05 | 7.82 ± 0.01 7.90 ± 0.10 | [7] | |
| F | 7.89 ± 0.01 7.85 ± 0.07 | 8.09 ± 0.01 8.15 ± 0.11 | 8.77 ± 0.01 8.71 ± 0.10 | [3] |
2.2. NMR Characterization
2.3. Molecular Structure and Conformation
2.4. Supramolecular Structure
2.5. Biological Activity
Docking Studies
3. Experimental
3.1. Materials
3.2. pKa Determination
3.3. Stability Studies
3.4. NMR Spectroscopy
3.5. Crystal Structure Determination
3.6. Theoretical Calculations
3.7. Biological Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gozdalik, J.T.; Adamczyk-Woźniak, A.; Sporzyński, A. Influence of fluorine substituents on the properties of phenylboronic compounds. Pure Appl. Chem. 2018, 90, 677–702. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Jakubczyk, M.; Sporzyński, A.; Żukowska, G. Quantitative determination of the Lewis acidity of phenylboronic catechol esters—Promising anion receptors for polymer electrolytes. Inorg. Chem. Commun. 2011, 14, 1753–1755. [Google Scholar] [CrossRef]
- Zarzeczańska, D.; Adamczyk-Woźniak, A.; Kulpa, A.; Ossowski, T.; Sporzyński, A. Fluorinated Boronic Acids: Acidity and Hydrolytic Stability of Fluorinated Phenylboronic Acids. Eur. J. Inorg. Chem. 2017, 2017, 4493–4498. [Google Scholar] [CrossRef] [Green Version]
- Cox, P.A.; Reid, M.; Leach, A.G.; Campbell, A.D.; King, E.J.; Lloyd-Jones, G.C. Base-Catalyzed Aryl-B(OH)2 Protodeboronation Revisited: From Concerted Proton Transfer to Liberation of a Transient Aryl Anion. J. Am. Chem. Soc. 2017, 139, 13156–13165. [Google Scholar] [CrossRef] [PubMed]
- Madura, I.; Czerwińska, K.; Sołdańska, D. Hydrogen-Bonded Dimeric Synthon of Fluoro-Substituted Phenylboronic Acids versus Supramolecular Organization in Crystals. Cryst. Growth Des. 2014, 14, 5912–5921. [Google Scholar] [CrossRef]
- Madura, I.D.; Czerwińska, K.; Jakubczyk, M.; Pawełko, A.; Adamczyk-Woźniak, A.; Sporzyński, A. Weak C-H⋯O and dipole-dipole interactions as driving forces in crystals of fluorosubstituted phenylboronic catechol esters. Cryst. Growth Des. 2013, 13, 5344–5352. [Google Scholar] [CrossRef]
- Gozdalik, J.T.; Marek, P.H.; Madura, I.D.; Gierczyk, B.; Popenda, Ł.; Schroeder, G.; Adamczyk-Woźniak, A.; Sporzyński, A. Structures and properties of trifluoromethylphenylboronic acids. J. Mol. Struct. 2019, 1180, 237–243. [Google Scholar] [CrossRef]
- Gierczyk, B.; Kaźmierczak, M.; Popenda, Ł.; Sporzyński, A.; Schroeder, G.; Jurga, S. Influence of fluorine substituents on the NMR properties of phenylboronic acids. Magn. Reson. Chem. 2014, 52, 202–213. [Google Scholar] [CrossRef]
- Kowalska, K.; Adamczyk-Woźniak, A.; Gajowiec, P.; Gierczyk, B.; Kaczorowska, E.; Popenda, Ł.; Schroeder, G.; Sikorski, A.; Sporzyński, A. Fluoro-substituted 2-formylphenylboronic acids: Structures, properties and tautomeric equilibria. J. Fluor. Chem. 2016, 187, 1–8. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Gozdalik, J.T.; Wieczorek, D.; Madura, I.D.; Kaczorowska, E.; Brzezińska, E.; Sporzyński, A.; Lipok, J. Synthesis, Properties and Antimicrobial Activity of 5-Trifluoromethyl-2-formylphenylboronic Acid. Molecules 2020, 25, 799. [Google Scholar] [CrossRef] [Green Version]
- Borys, K.M.; Wieczorek, D.; Pecura, K.; Lipok, J.; Adamczyk-Woźniak, A. Antifungal activity and tautomeric cyclization equilibria of formylphenylboronic acids. Bioorg. Chem. 2019, 91, a103081. [Google Scholar] [CrossRef] [PubMed]
- Siodła, T.; Ozimiński, W.P.; Hoffmann, M.; Koroniak, H.; Krygowski, T.M. Toward a Physical Interpretation of Substituent Effects: The Case of Fluorine and Trifluoromethyl Groups. J. Org. Chem. 2014, 79, 7321–7331. [Google Scholar] [CrossRef]
- Castagnetti, E.; Schlosser, M. 2-, 3-, and 4-(Trifluoromethoxy)phenyllithiums: Versatile Intermediates Offering Access to a Variety of New Organofluorine Compounds. Eur. J. Org. Chem. 2001, 2001, 691–695. [Google Scholar] [CrossRef]
- Leermann, T.; Leroux, F.R.; Colobert, F. Highly Efficient One-Pot Access to Functionalized Arylboronic Acids via Noncryogenic Bromine/Magnesium Exchanges. Org. Lett. 2011, 13, 4479–4481. [Google Scholar] [CrossRef] [PubMed]
- Mfuh, A.M.; Doyle, J.D.; Chhetri, B.; Arman, H.D.; Larionov, O.V. Scalable, Metal- and Additive-Free, Photoinduced Borylation of Haloarenes and Quaternary Arylammonium Salts. J. Am. Chem. Soc. 2016, 138, 2985–2988. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zuo, J.; Tang, W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front. Pharmacol. 2018, 9, a1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Wu, S.; Yan, S.; Li, C.; Abduhulam, H.; Shi, Y.; Dang, Y.; Cao, C. Suzuki–Miyaura Cross-Coupling of Sulfoxides. ACS Catal. 2020, 10, 8168–8176. [Google Scholar] [CrossRef]
- Cao, Z.; Zhu, Y.; Li, X.; He, Y.; Zhang, J.; Xu, L.; Wei, Y. tert-Butyl Bromide-Promoted Intramolecular Cyclization of 2-Arylamino Phenyl Ketones and Its Combination with Cu-Catalyzed C–N Coupling: Synthesis of Acridines at Room Temperature. J. Org. Chem. 2020, 85, 10167–10174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Jia, Q.; Wang, Y.; Ma, Y.; Szostak, M. Ruthenium (II)-Catalyzed C–H Arylation of N,N-Dialkyl Thiobenzamides with Boronic Acids by Sulfur Coordination in 2-MeTHF. Org. Lett. 2020, 22, 6884–6890. [Google Scholar] [CrossRef]
- Dutta, S.; Yang, S.; Vanjari, R.; Mallick, R.K.; Gandon, V.; Sahoo, A.K. Keteniminium-Driven Umpolung Difunctionalization of Ynamides. Angew. Chem. Int. Ed. 2020, 59, 10785–10790. [Google Scholar] [CrossRef]
- Kancharla, P.; Dodean, R.A.; Li, Y.; Pou, S.; Pybus, B.; Melendez, V.; Read, L.; Bane, C.E.; Vesely, B.; Kreishman-Deitrick, M.; et al. Lead Optimization of Second-Generation Acridones as Broad-Spectrum Antimalarials. J. Med. Chem. 2020, 63, 6179–6202. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hu, X.-D.; Zhang, H.; Zhang, X.-W.; Cai, J.; Usman, M.; Cong, H.; Liu, W.-B. Enantioselective Assembly of Cycloenones with a Nitrile-Containing All-Carbon Quaternary Center from Malononitriles Enabled by Ni Catalysis. J. Am. Chem. Soc. 2020, 142, 7328–7333. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Peng, X.; Li, C.-J.; Zeng, H. Photoinduced catalyst-free deborylation–deuteration of arylboronic acids with D2O. Green Chem. 2020, 22, 6323–6327. [Google Scholar] [CrossRef]
- Planas, O.; Peciukenas, V.; Cornella, J. Bismuth-Catalyzed Oxidative Coupling of Arylboronic Acids with Triflate and Nonaflate Salts. J. Am. Chem. Soc. 2020, 142, 11382–11387. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Q.; Rong, J.; Ren, J.; Song, X.; Fan, X.; Shen, M.; Xia, Y.; Wang, N.; Liu, Z.; et al. Synthesis and biological evaluation of (1,2,4)triazole [4,3-a]pyridine derivatives as potential therapeutic agents for concanavalin A-induced hepatitis. Eur. J. Med. Chem. 2019, 179, 182–195. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Zhang, L.; Chen, D.; Dong, Z.; Zhao, C.; Liu, Z.; Xia, Q.; Wu, J.; Chen, Y.; et al. Design, synthesis, and biological evaluation of indazole derivatives as selective and potent FGFR4 inhibitors for the treatment of FGF19-driven hepatocellular cancer. Eur. J. Med. Chem. 2021, 214, 113219. [Google Scholar] [CrossRef]
- Sun, N.; Huang, Y.; Yu, M.; Zhao, Y.; Chen, J.-A.; Zhu, C.; Song, M.; Guo, H.; Xie, Q.; Wang, Y. Discovery of carboxyl-containing biaryl ureas as potent RORγt inverse agonists. Eur. J. Med. Chem. 2020, 202, 112536. [Google Scholar] [CrossRef]
- Jurrat, M.; Maggi, L.; Lewis, W.; Ball, L.T. Modular bismacycles for the selective C–H arylation of phenols and naphthols. Nat. Chem. 2020, 12, 260–269. [Google Scholar] [CrossRef]
- Hansen, J.S.; Christensen, J.B.; Solling, T.I.; Jakobsen, P.; Hoeg-Jensen, T. Ortho-substituted aryl monoboronic acids have improved selectivity for d-glucose relative to d-fructose and l-lactate. Tetrahedron 2011, 67, 1334–1340. [Google Scholar] [CrossRef]
- Golovanov, I.S.; Mazeina, G.S.; Nelyubina, Y.V.; Novikov, R.A.; Mazur, A.S.; Britvin, S.N.; Tartakovsky, V.A.; Ioffe, S.L.; Sukhorukov, A.Y. Exploiting Coupling of Boronic Acids with Triols for a pH-Dependent “Click-Declick” Chemistry. J. Org. Chem. 2018, 83, 9756–9773. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Borys, K.M.; Czerwińska, K.; Gierczyk, B.; Jakubczyk, M.; Madura, I.D.; Sporzyńki, A.; Tomecka, E. Intramolecular interactions in ortho-methoxyalkylphenylboronic acids and their catechol esters. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 116, 616–621. [Google Scholar] [CrossRef]
- Kajimoto, O.; Saeki, T.; Nagaoka, Y.; Fueno, T. Temperature-jump rate studies of the association reactions of boric and benzeneboronic acids with hydroxide ion. J. Phys. Chem. 1977, 81, 1712–1716. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Sporzyński, A. The influence of ortho-substituents on the properties of phenylboronic acids. J. Organomet. Chem. 2020, 913, a121202. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Cyrański, M.K.; Dąbrowska, A.; Gierczyk, B.; Klimentowska, P.; Schroeder, G.; Żubrowska, A.; Sporzyński, A. Hydrogen bonds in phenylboronic acids with polyoxaalkyl substituents at ortho-position. J. Mol. Struct. 2009, 920, 430–435. [Google Scholar] [CrossRef]
- Durka, K.; Jarzembska, K.N.; Kamiński, R.; Luliński, S.; Serwatowski, J.; Woźniak, K. Structural and Energetic Landscape of Fluorinated 1,4-Phenylenediboronic Acids. Cryst. Growth Des. 2012, 12, 3720–3734. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, Y.A. On the Possibility of Kinetic Energy Density Evaluation from the Experimental Electron-Density Distribution. Acta Crystallogr. Sect. A 1997, 53, 264–272. [Google Scholar] [CrossRef]
- Cyrański, M.K.; Klimentowska, P.; Rydzewska, A.; Serwatowski, J.; Sporzyński, A.; Stępień, D.K. Towards a monomeric structure of phenylboronic acid: The influence of ortho-alkoxy substituents on the crystal structure. CrystEngComm 2012, 14, 6282–6294. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park, KS, USA. 2019. Available online: http://aim.tkgristmill.com/ (accessed on 30 March 2021).
- Sheldrick, G. A Short History of ShelX. Acta Crystallogr. A. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimpi, M.R.; SeethaLekshmi, N.; Pedireddi, V.R. Supramolecular Architecture in Some 4-Halophenylboronic Acids. Cryst. Growth Des. 2007, 7, 1958–1963. [Google Scholar] [CrossRef]
- Cyrański, M.K.; Jezierska, A.; Klimentowska, P.; Panek, J.J.; Sporzyński, A. Impact of intermolecular hydrogen bond on structural properties of phenylboronic acid: Quantum chemical and X-ray study. J. Phys. Org. Chem. 2008, 21, 472–482. [Google Scholar] [CrossRef]
- Takamizawa, S.; Takasaki, Y.; Sasaki, T.; Ozaki, N. Superplasticity in an organic crystal. Nat. Commun. 2018, 9, a3984. [Google Scholar] [CrossRef]
- Palencia, A.; Crépin, T.; Vu, M.T.; Lincecum, T.L.; Martinis, S.A.; Cusack, S. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2012, 19, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Kostrowicki, J.; Liwo, A. Determination of equilibrium parameters by minimization of an extended sum of squares. Talanta 1990, 37, 645–650. [Google Scholar] [CrossRef]
- Kostrowicki, J.; Liwo, A. DECFAM—A new computer oriented algorithm for the determination of equilibrium constants from potentionmetric and/or spectrophotometric measurements—I: Basic principles of the method and calculations of equilibrium concentrations. Comput. Chem. 1984, 8, 91–99. [Google Scholar] [CrossRef]
- Kostrowicki, J.; Liwo, A. DECFAM—A new computer oriented algorithm for the determination of equilibrium constants from potentiometric and/or spectrophotometric measurements—II: Methods based on analytical expressions. Comput. Chem. 1984, 8, 101–105. [Google Scholar] [CrossRef]
- Chylewska, A.; Jacewicz, D.; Zarzeczańska, D.; Chmurzyński, L. Determination of dissociation constants for coordination compounds of Cr (III) and Co (III) using potentiometric and spectrophotometric methods. J. Chem. Thermodyn. 2008, 40, 1290–1294. [Google Scholar] [CrossRef]
- Crysalis Pro. Version 1.171.38.46, Rigaku; Oxford Diffraction: Abingdon-on-Thames, UK, 2015.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]








| B(OH)2 | H2 | H3 | H4 | H5 | H6 | |
|---|---|---|---|---|---|---|
| 1 | 7.33 (s) | - | 7.31–7.25 (m) | 7.50 (ddd) 3JH-H = 12.5 Hz 3JH-H = 8.2 Hz 4JH-H = 1.8 Hz | 7.40–7.32 (m) overlapping with B(OH)2 signal * | 7.76 (dd) 3JH-H = 7.3 Hz 4JH-H = 1.8 Hz |
| 2 | 7.64 (s) | 7.79–7.72 (m) | - | 7.40–7.31 (m) | 7.53–7.44 (m) | 7.87 (ddd) 3JH-H = 7.3 Hz 4JH-H = 1.8 Hz 4JH-H = 1 Hz |
| 3 | 7.32 (s) | 8.08–7.94 (m) | 7.34–7.24 (m) | - | 7.34–7.24 (m) | 8.08–7.94 (m) |
| OCF3 | C1 | C2 | C3 | C4 | C5 | C6 | |
|---|---|---|---|---|---|---|---|
| 1 | 121.5 (q) 1JC-F = 254.7 Hz | n.d. | 153.3 (q) 3JC-F = 1.7 Hz | 121.5 (q) 4JC-F = 1.0 Hz | 132.2 (s) | 127.5 (s) | 136.4 (s) |
| 2 | 121.4 (q) 1JC-F = 255.0 Hz | n.d. | 126.8 (s) | 149.7 (q) 3JC-F = 1.7 Hz | 123.6 (s) | 130.2 (s) | 133.7 (s) |
| 3 | 121.4 (q) 1JC-F = 255.5 Hz | n.d. | 137.0 (s) | 120.6 (s) | 151.7 (q) 3JC-F = 1.7 Hz | 120.6 (s) | 137.0 (s) |
| 11B-NMR | 19F-NMR | |
|---|---|---|
| 1 | 28 (s) | −56.93 (d), 5JH3-F = 1.4 Hz |
| 2 | 28 (s) | −57.64 (dd), 5JH2-F = 0.9 Hz, 5JH4-F = 1 Hz |
| 3 | 28 (s) | −57.59 (t) 5JH3H5-F = 1 Hz |
| 1 | 3-A a | 3-B a | |
|---|---|---|---|
| dC1-B1/Å | 1.578(2) | 1.569(4) | 1.565(4) |
| dO1-B1/Å | 1.365(2) | 1.364(4) | 1.359(3) |
| dO2-B1/Å | 1.365(2) | 1.358(3) | 1.365(4) |
| /Å | /Å | ρ(rCP)/e∙A−3 | L(rCP) /e∙A−5 | EELint/kJ mol−1 | |||
|---|---|---|---|---|---|---|---|
| 1 | 0.961/0.839 | 2.081/2.377 | 2.817/2.839(2) | 132.10/115.3(1) | 0.136 | 0.471 | 19.0 |
| 1a | 0.964/0.859 [42] | 1.924/1.936 | 2.719/2.654 | 138.10/140.22 | 0.177 | 0.655 | 29.3 |
| Donor… Acceptor | Energy kJ mol−1 | |
|---|---|---|
| 1 | LP1(O) …BD3*(C-F1) | 6.5 |
| LP1(O) …BD*(Car-Car) | 4.8 | |
| LP2(O) …BD1*(C-F2) | 16.5 | |
| LP2(O) …BD2*(C-F3) | 15.1 | |
| 1a | LP1(O) …BD3*(C-H1) | 3.3 |
| LP1(O) …BD*(Car-Car) | 6.6 | |
| LP2(O) …BD1*(C-H2) | 6.5 | |
| LP2(O) …BD2*(C-H3) | 6.4 |
| QTAIM | NBO | ||||||
|---|---|---|---|---|---|---|---|
| L(r) (3, −3) | ρ(rCP)/e∙A−3 | L(rCP) /e∙A−5 | Orbital | Occupancy/e | Orbital | Occupancy/e | |
| 1 | CC1(O) | 6.468 | 125.5 | LP1(O) | 1.900 | BD*(O–H) | 0.011 |
| CC2 (O) | 6.379 | 125.8 | LP2(O) | 1.935 | BD1*(C–F) BD2*(C–F) BD3*(C–F) | 0.111 0.107 0.091 | |
| 1a | CC1 (O) | 6.479 | 121.3 | LP1(O) | 1.868 | BD*(O–H) | 0.016 |
| CC2 (O) | 6.379 | 121.2 | LP2(O) | 1.957 | BD1*(C–H) BD2*(C–H) BD3*(C–H) | 0.017 0.017 0.008 |
| Lowest Binding Energy (kcal mol−1) | Mean Binding Energy (kcal mol−1) | Number in Cluster | Inhibition Constant (μM) | Number of H-Bonds | |
|---|---|---|---|---|---|
| 1 | −4.65 | −4.45 | 73 | 391.52 | 4 |
| 2 | −4.73 | −4.59 | 36 | 341.78 | 3 |
| 3 | −5.29 | −4.92 | 94 | 132.74 | 5 |
| 1-AMP | −7.32 | −6.91 | 9 | 4.30 | 2 |
| 2-AMP | −7.07 | −6.77 | 6 | 6.62 | 1 |
| 3-AMP | −6.84 | −6.14 | 22 | 9.70 | 3 |
| 10 μg | 25 μg | 50 μg | 100 μg | DMSO | MIC (μg/mL) | |
|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0 | 0 | >500 |
| 2 | 0 | 0 | 0 | 1.7 ± 1.5 | 0 | 250 |
| 3 | 0 | 0 | 0 | 1.3 ± 0.6 | 0 | 250 |
| Streptomycin | 13 ± 1 | 14.7 ± 0.6 | 15.7 ± 0.6 | 16 ± 0 | 0 | 2 |
| 10 μg | 25 μg | 50 μg | 100 μg | DMSO | MIC (μg/mL) | |
|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0 | 0 | >500 |
| 2 | 0 | 0 | 6 ± 1 | 11 ± 1 | 0 | 125 |
| 3 | 0 | 5 ± 2 | 8 ± 1 | 12 ± 1 | 0 | 125 |
| Streptomycin | 16 ± 1 | 19 ± 1 | 20 ± 1 | 23 ± 1 | 0 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamczyk-Woźniak, A.; Gozdalik, J.T.; Kaczorowska, E.; Durka, K.; Wieczorek, D.; Zarzeczańska, D.; Sporzyński, A. (Trifluoromethoxy)Phenylboronic Acids: Structures, Properties, and Antibacterial Activity. Molecules 2021, 26, 2007. https://doi.org/10.3390/molecules26072007
Adamczyk-Woźniak A, Gozdalik JT, Kaczorowska E, Durka K, Wieczorek D, Zarzeczańska D, Sporzyński A. (Trifluoromethoxy)Phenylboronic Acids: Structures, Properties, and Antibacterial Activity. Molecules. 2021; 26(7):2007. https://doi.org/10.3390/molecules26072007
Chicago/Turabian StyleAdamczyk-Woźniak, Agnieszka, Jan T. Gozdalik, Ewa Kaczorowska, Krzysztof Durka, Dorota Wieczorek, Dorota Zarzeczańska, and Andrzej Sporzyński. 2021. "(Trifluoromethoxy)Phenylboronic Acids: Structures, Properties, and Antibacterial Activity" Molecules 26, no. 7: 2007. https://doi.org/10.3390/molecules26072007
APA StyleAdamczyk-Woźniak, A., Gozdalik, J. T., Kaczorowska, E., Durka, K., Wieczorek, D., Zarzeczańska, D., & Sporzyński, A. (2021). (Trifluoromethoxy)Phenylboronic Acids: Structures, Properties, and Antibacterial Activity. Molecules, 26(7), 2007. https://doi.org/10.3390/molecules26072007