
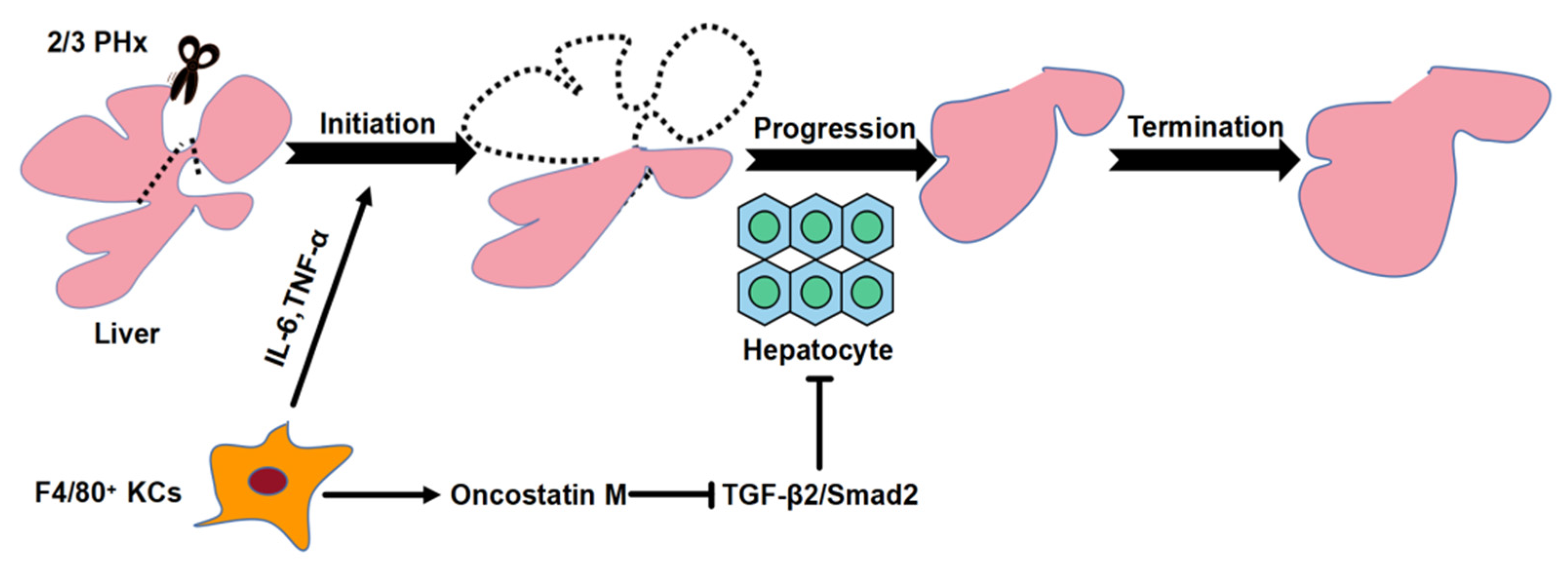
F4/80+ Kupffer Cell-Derived Oncostatin M Sustains the Progression Phase of Liver Regeneration through Inhibition of TGF-β2 Pathway

 ,
, 
Abstract
:1. Introduction
2. Results
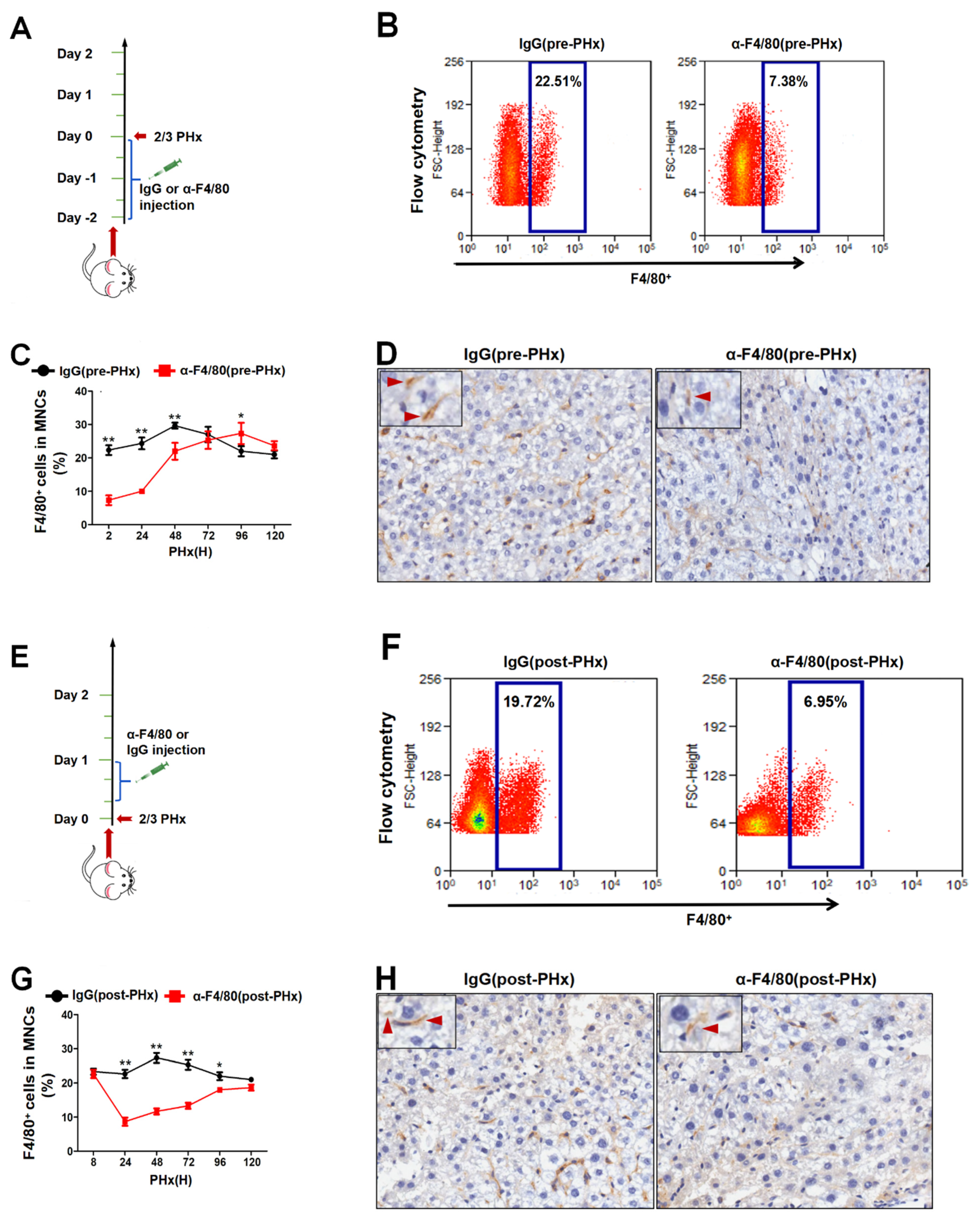
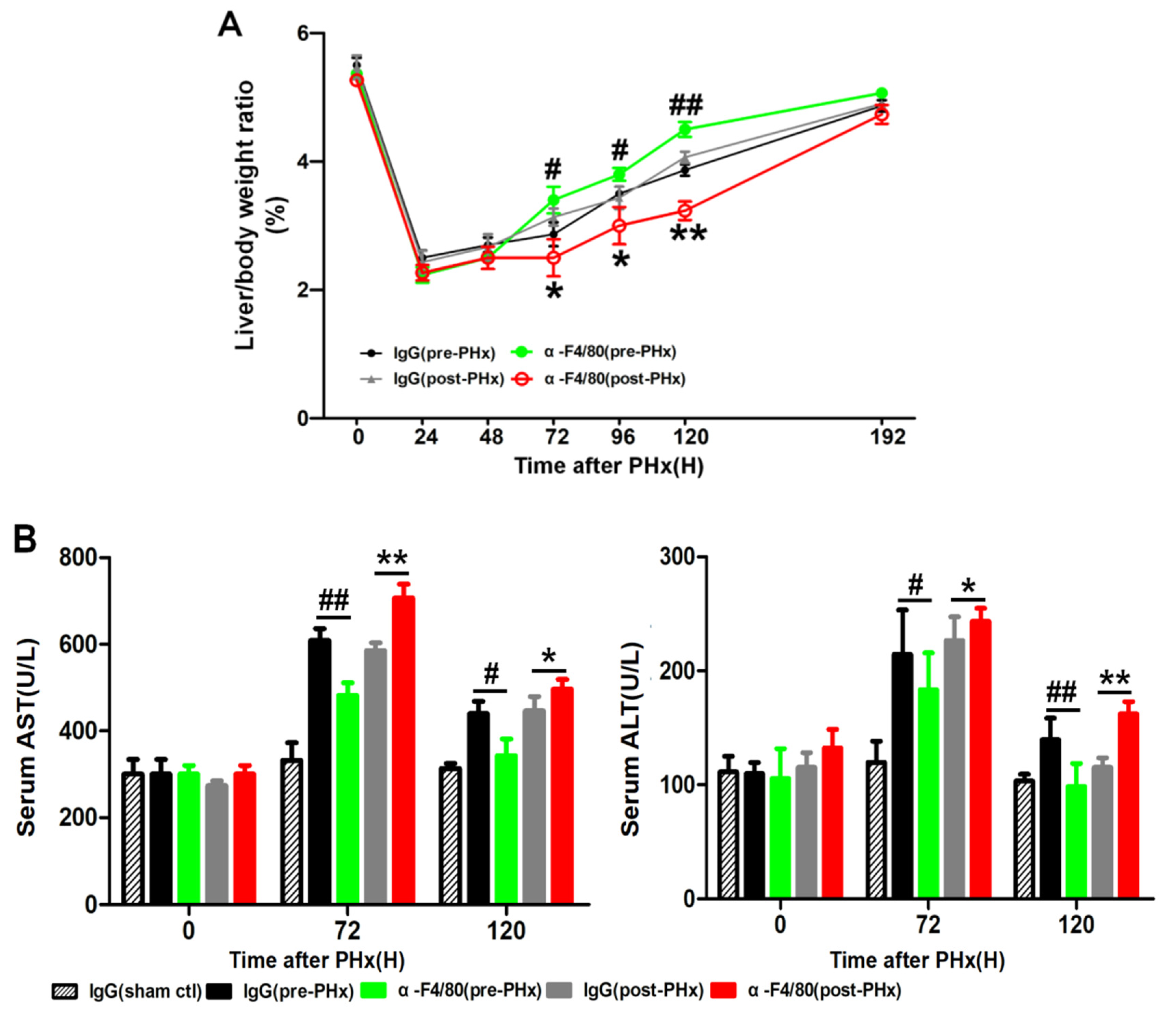
2.1. F4/80+ KCs Play a Dual Role in Controlling the Initiation and the Progression of Liver Regeneration Rather Than the Termination
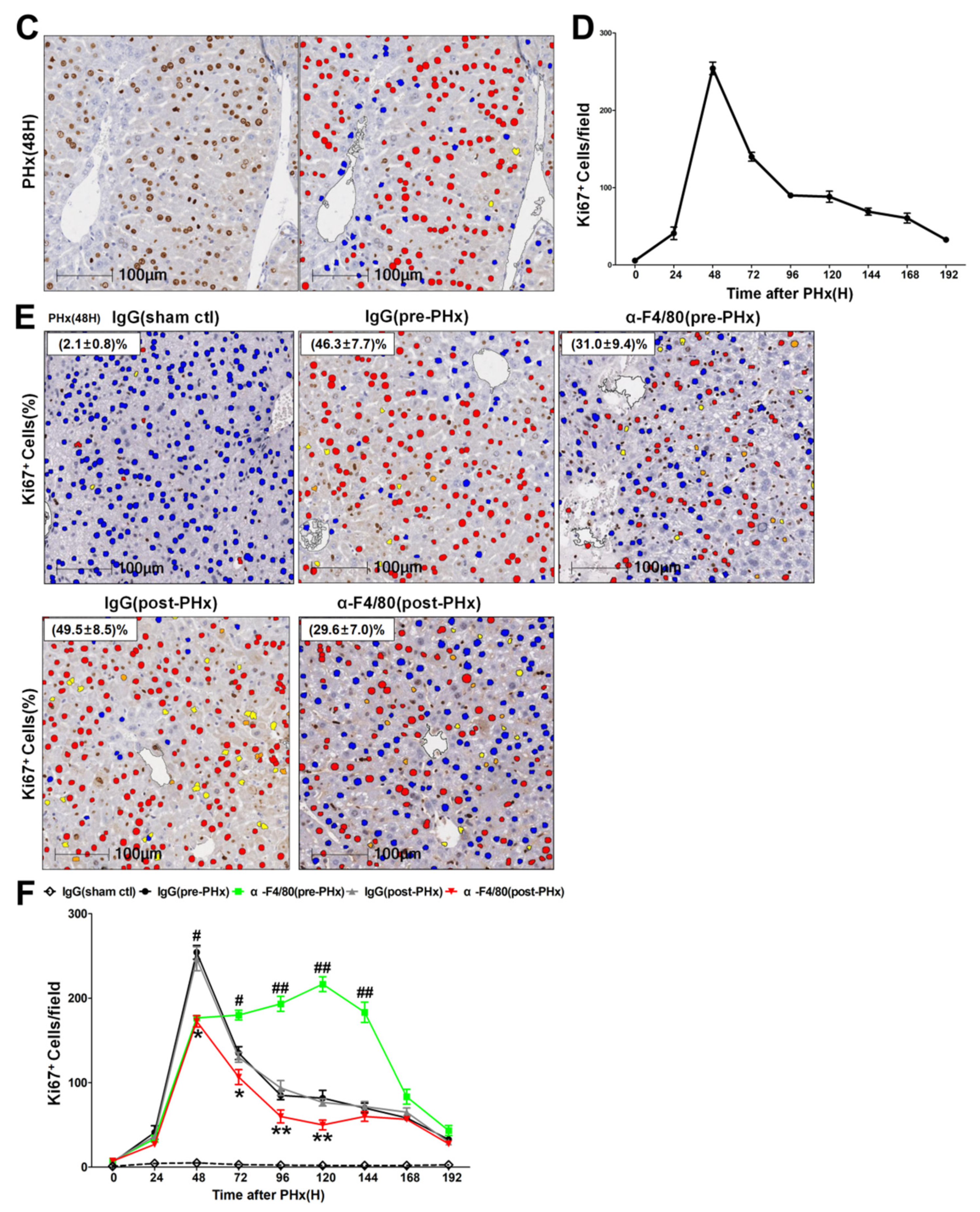
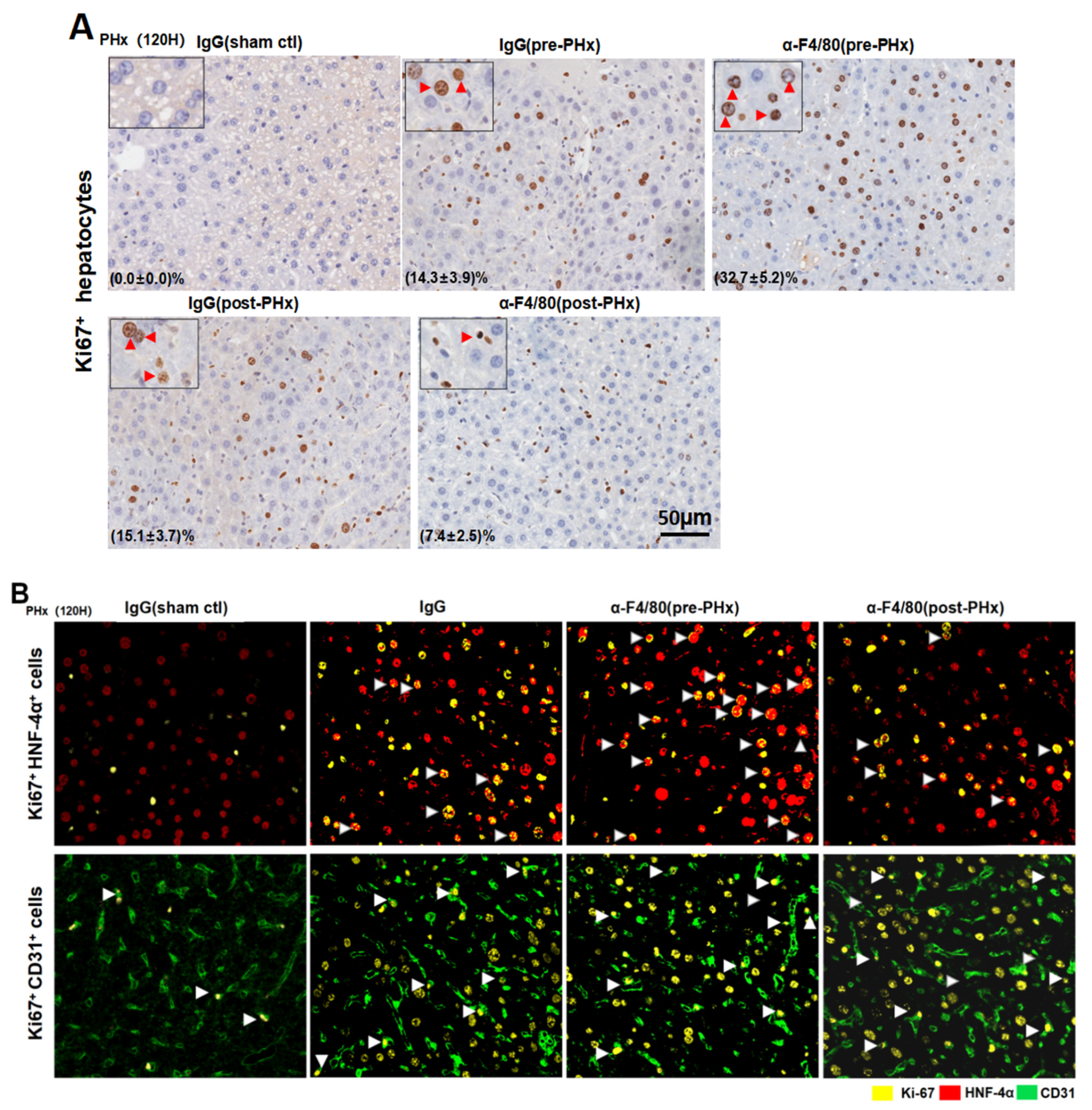
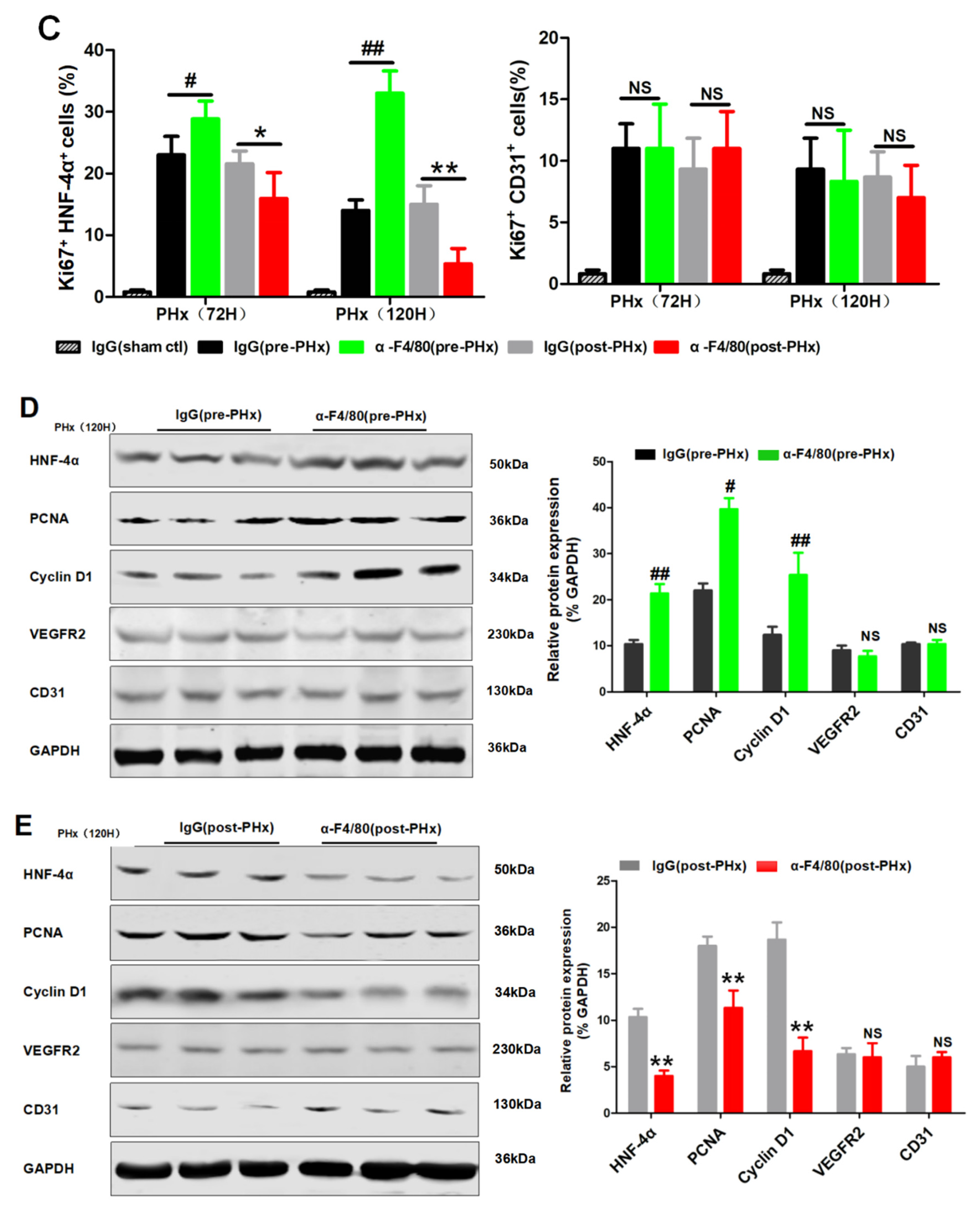
2.2. F4/80+ KCs Maintain Hepatocyte Proliferation during the Progression Phase of Liver Regeneration
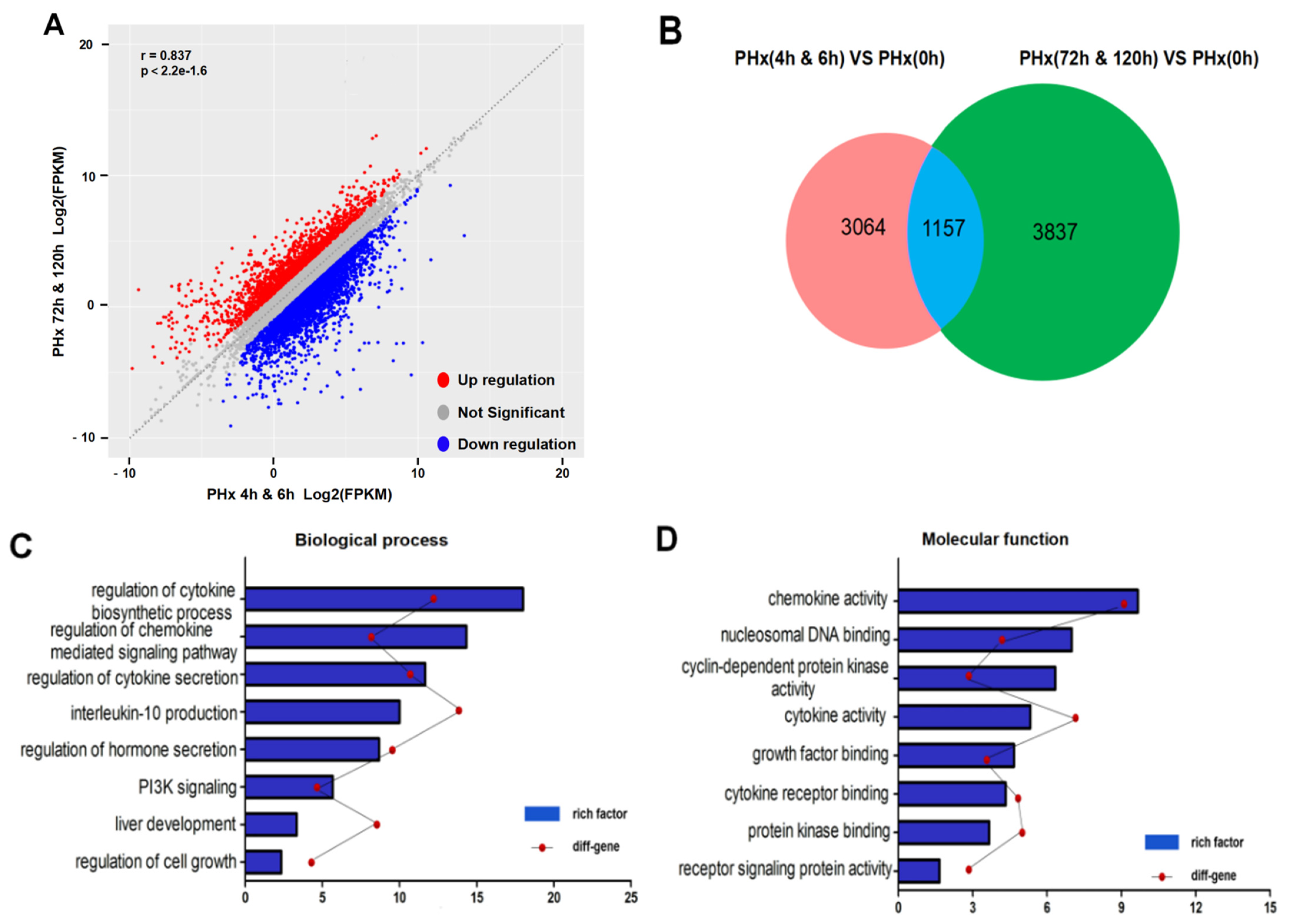
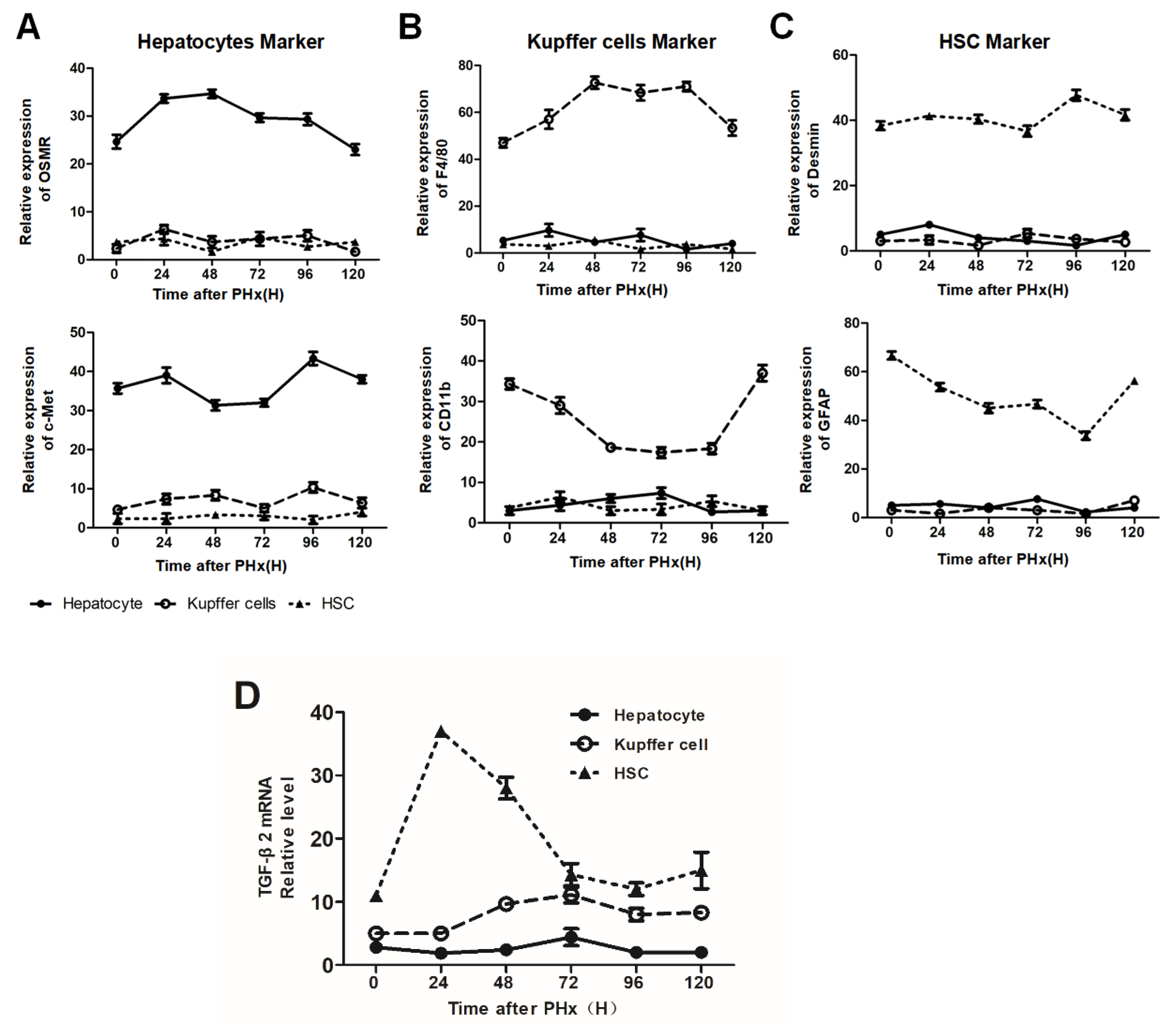
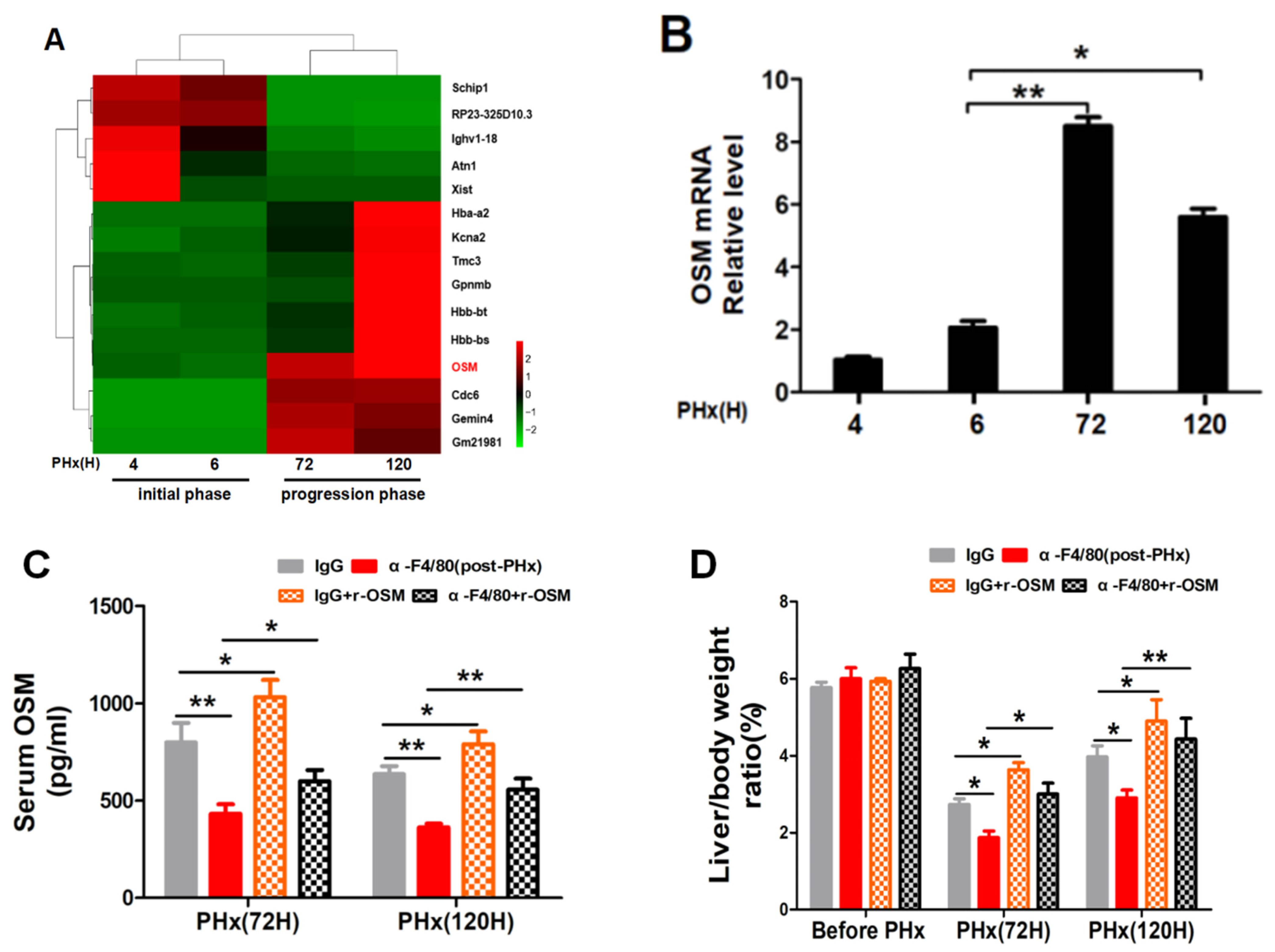
2.3. Different Transcriptional Alterations Occur in F4/80+ KCs between the Initiation and Progression Stages of Liver Regeneration
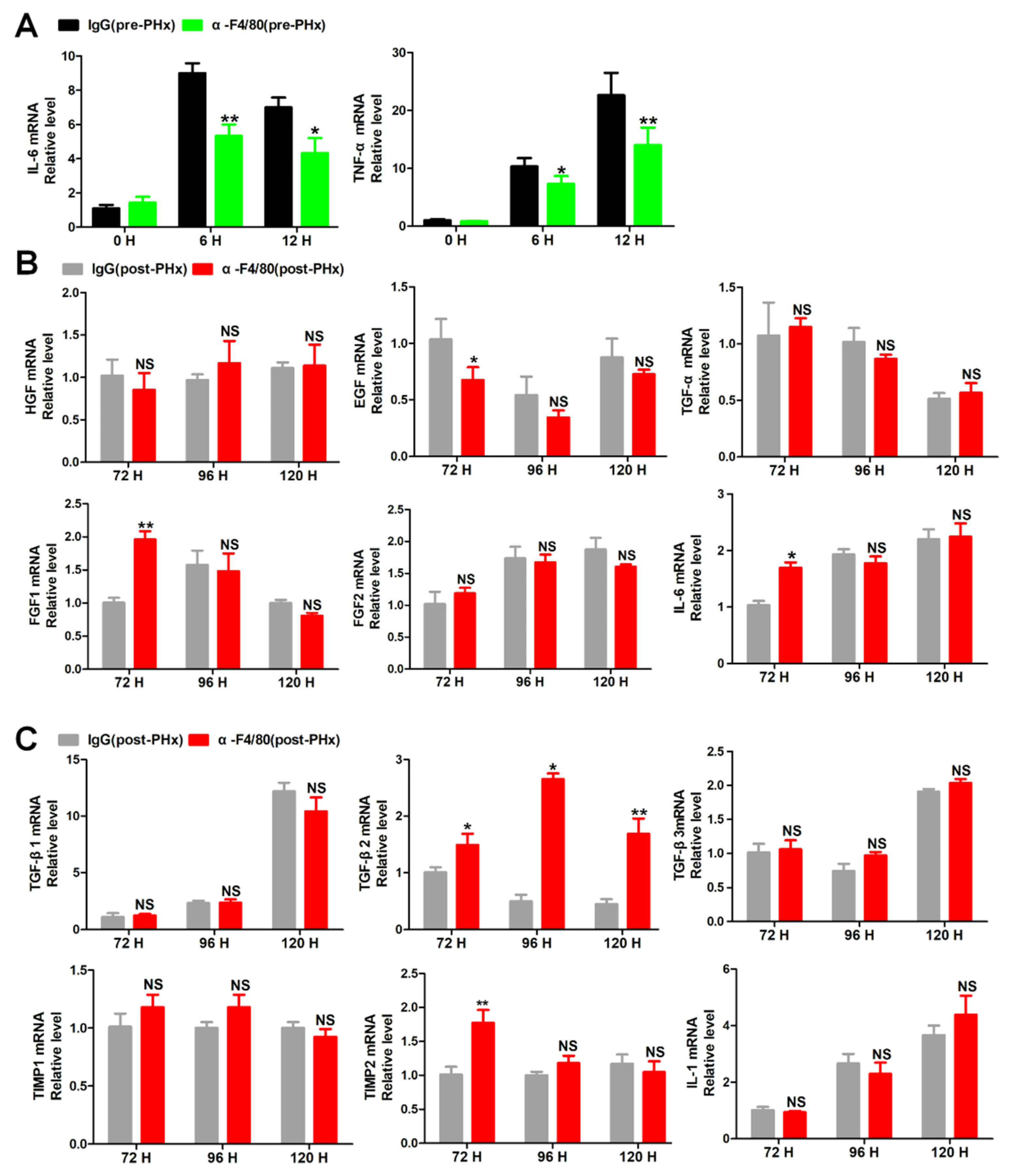
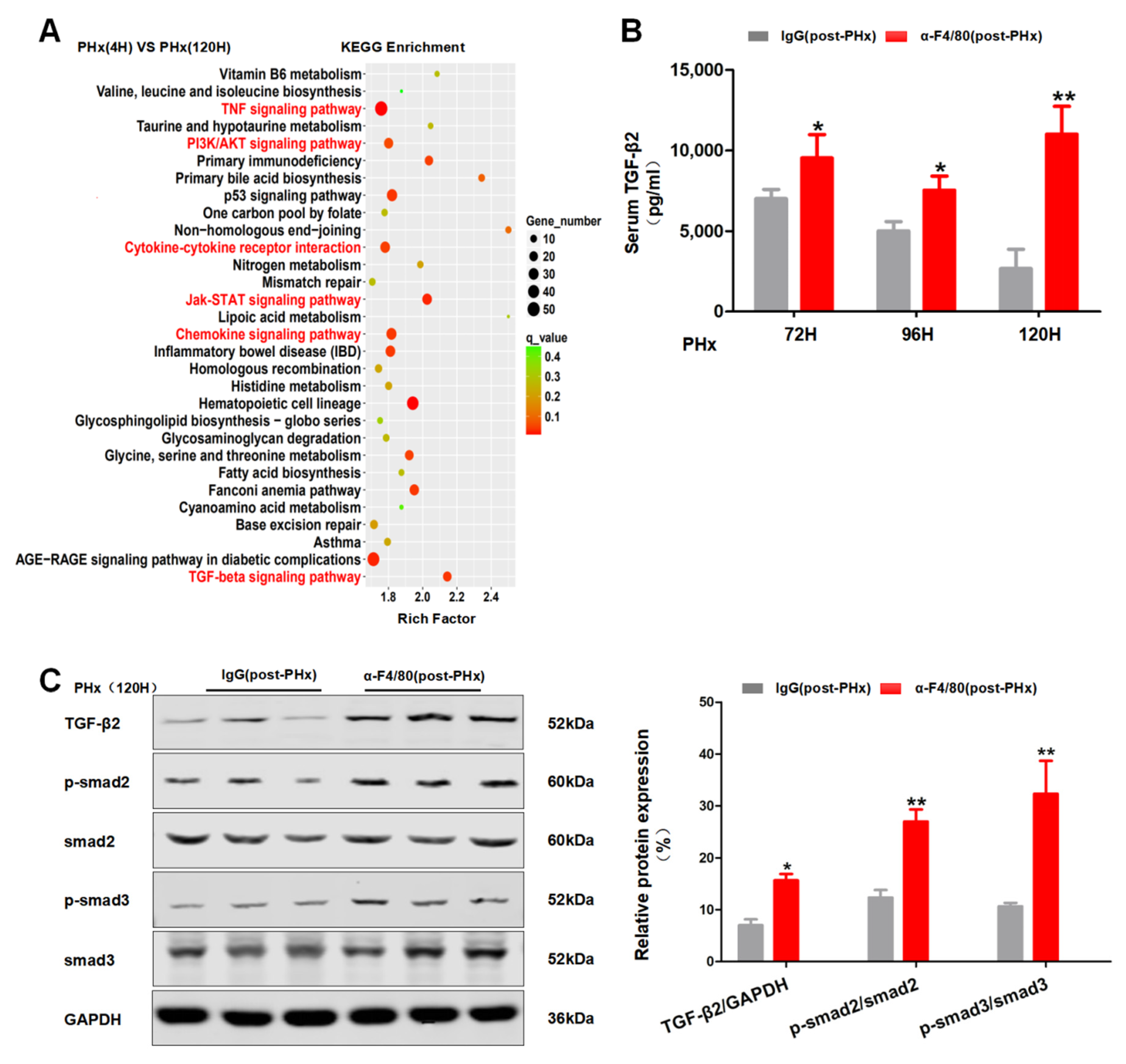
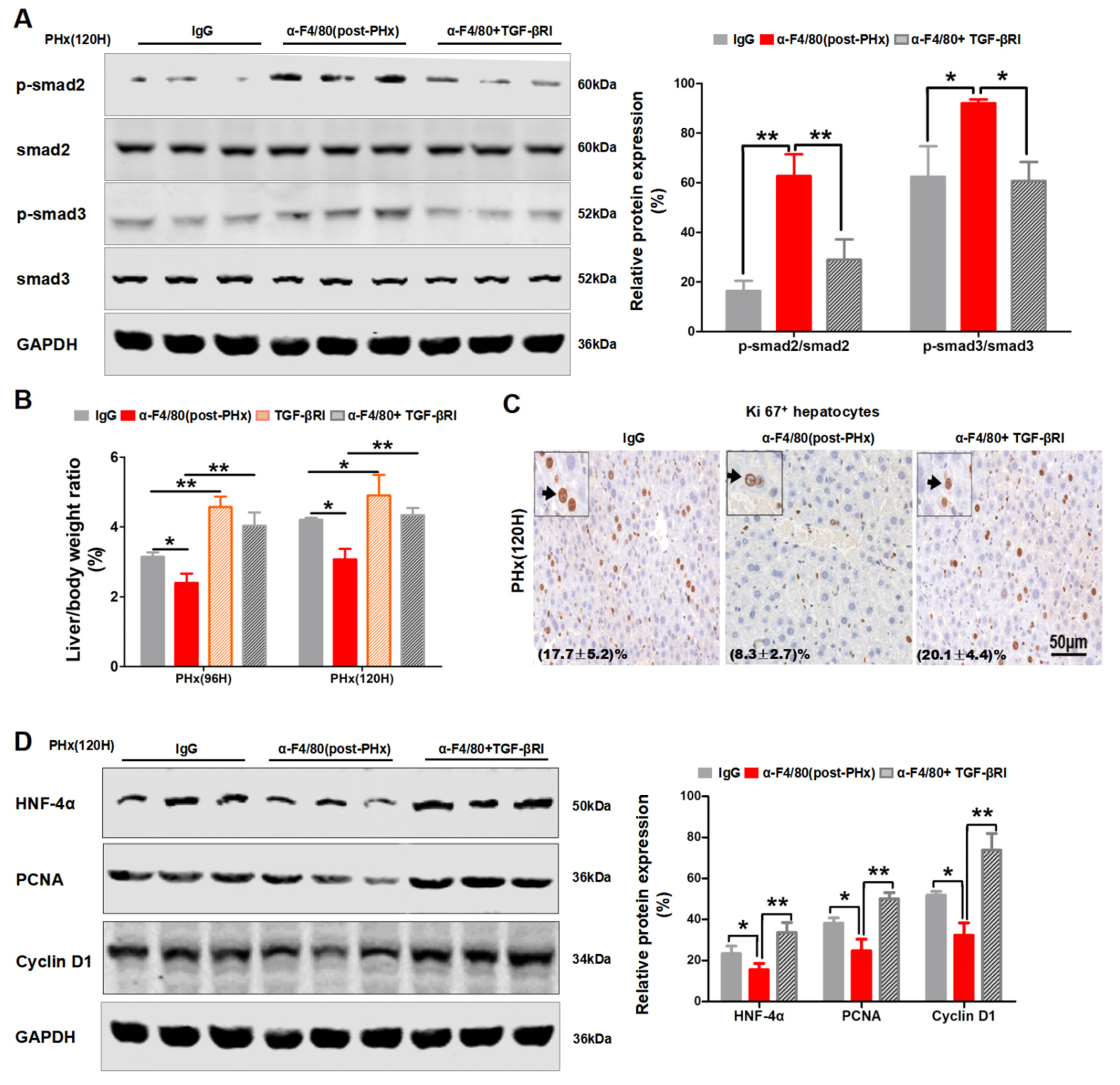
2.4. F4/80+ KCs Maintain the Progression of Liver Regeneration through Inhibition of the TGF-β2/smad2 Pathway
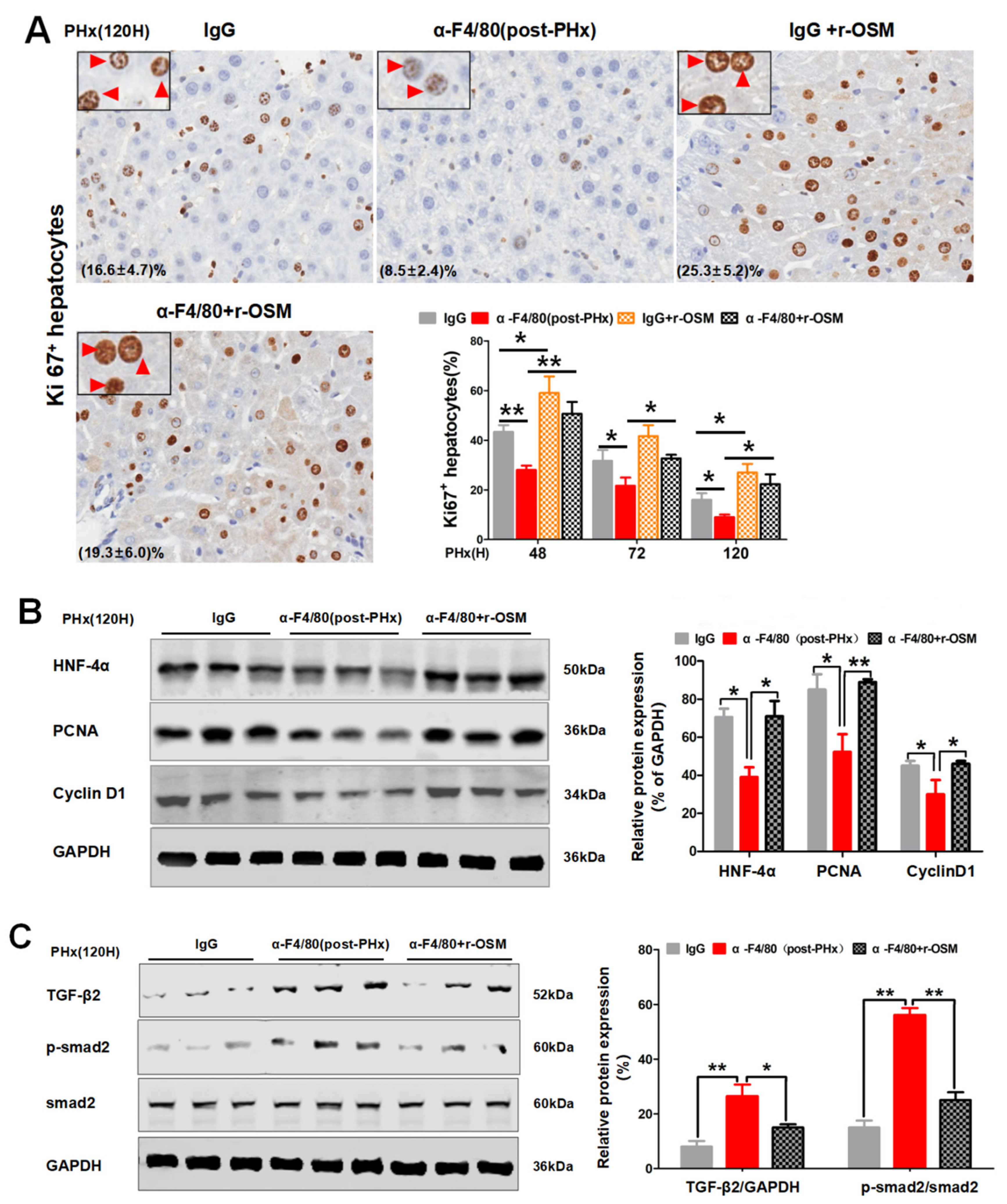
2.5. F4/80+ KC-Derived OSM Inhibits the TGF-β2/smad2 Pathway to Drive the Progression Phase of Liver Regeneration
3. Discussion
4. Methods
4.1. Animal Treatments and 2/3 PHx
4.2. Isolation of Primary Hepatocytes and Nonparenchymal Cells (NPCs)
4.3. Library Construction for RNA-Seq and Sequencing Procedures
4.4. Immunofluorescence and Immunohistochemical (IHC) Staining
4.5. Digital Image Analysis of Ki67 with Virtual Dual Staining
4.6. Real-Time PCR
4.7. Western Blotting
4.8. Measurement of the Serum ALT, AST, TGF-β2, and OSM
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Higgins, G.M.; Anderson, R.M. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch. Pathol. 1931, 12, 186–202. [Google Scholar]
- Nygard, I.E.; Mortensen, K.E.; Hedegaard, J.; Conley, L.N.; Kalstad, T.; Bendixen, C.; Revhaug, A. The genetic regulation of the terminating phase of liver regeneration. Comp. Hepatol. 2012, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.J.; Newsome, P.N. Liver regeneration-mechanisms and models to clinical application. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.I.; Mars, W.M.; Michalopoulos, G.K. Signals and cells involved in regulating liver regeneration. Cells 2012, 1, 1261–1292. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Zhang, C. Liver regeneration: Influence-factors and mechanism of mesenchymal stem cell transplantation. Trop. J. Pharm. Res. 2019, 17. [Google Scholar] [CrossRef]
- Kiyoshi, N.; Hiroyuki, N.; Masami, I.; Manabu, K.; Masahiro, N.; Suefumi, A.; Shuhji, S.; Junji, Y.; Schmidt, E.E. Mouse CD11b + Kupffer cells recruited from bone marrow accelerate liver regeneration after partial hepatectomy. PLoS ONE 2015, 10, e0136774. [Google Scholar]
- Boulton, R.A.; Alison, M.R.; Golding, M.; Selden, C.; Hodgson, H.J. Augmentation of the early phase of liver regeneration after 70% partial hepatectomy in rats following selective Kupffer cell depletion. J. Hepatol. 1998, 29, 271–280. [Google Scholar] [CrossRef]
- Hume, D.A.; Robinson, A.P.; MacPherson, G.G.; Gordon, S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Relationship between macrophages, Langerhans cells, reticular cells, and dendritic cells in lymphoid and hematopoietic organs. J. Exp. Med. 1983, 158, 1522–1536. [Google Scholar] [CrossRef]
- Kinoshita, M.; Uchida, T.; Sato, A.; Nakashima, M.; Nakashima, H.; Shono, S.; Habu, Y.; Miyazaki, H.; Hiroi, S.; Seki, S. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J. Hepatol. 2010, 53, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Albacker, L.A.; Yu, S.; Bedoret, D.; Lee, W.L.; Umetsu, S.E.; Monahan, S.; Freeman, G.J.; Umetsu, D.T.; Dekruyff, R.H. TIM-4, expressed by medullary macrophages, regulates respiratory tolerance by mediating phagocytosis of antigen-specific T cells. Mucosal Immunol. 2013, 6, 580–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoret, D.; Wallemacq, H.; Marichal, T.; Desmet, C.; Quesada Calvo, F.; Henry, E.; Closset, R.; Dewals, B.; Thielen, C.; Gustin, P.; et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Investig. 2009, 119, 3723–3738. [Google Scholar] [CrossRef] [PubMed]
- Van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abshagen, K.; Eipel, C.; Kalff, J.C.; Menger, M.D.; Vollmar, B. Loss of NF-kappaB activation in Kupffer cell-depleted mice impairs liver regeneration after partial hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1570–G1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.Y.; Ma, S.F.; Qu, J.F.; Tian, D.H. Effects of Kupffer cell inactivation on graft survival and liver regeneration after partial liver transplantation in rats. Hepatobil. Pancreat. Dis. Int. 2015, 14, 56–62. [Google Scholar] [CrossRef]
- Haldrup, D.; Heebøll, S.; Thomsen, K.L.; Andersen, K.J.; Grønbæk, H. Preserved liver regeneration capacity after partial hepatectomy in rats with non-alcoholic steatohepatitis. World J. Hepatol. 2018, 10, 8–21. [Google Scholar] [CrossRef]
- An, S.; Soe, K.; Akamatsu, M.; Hishikawa, Y.; Koji, T. Accelerated proliferation of hepatocytes in rats with iron overload after partial hepatectomy. Histochem. Cell Biol. 2012, 138, 773–786. [Google Scholar] [CrossRef]
- Lammert, E.; Axnick, J.; Buschmann, T. Endothelial cell-derived signals in liver development and regeneration. Eur. J. Med. Res. 2014, 19, S2. [Google Scholar] [CrossRef] [Green Version]
- Melgar-Lesmes, P.; Edelman, E.R. Monocyte-endothelial cell interactions in the regulation of vascular sprouting and liver regeneration in mouse. J. Hepatol. 2015, 63, 917. [Google Scholar] [CrossRef] [Green Version]
- Deleve, L.D. Liver sinusoidal endothelial cells and liver regeneration. J. Clin. Investig. 2013, 123, 1861–1866. [Google Scholar] [CrossRef]
- Hoffmann, K.; Nagel, A.J.; Tanabe, K.; Fuchs, J.; Dehlke, K.; Ghamarnejad, O.; Lemekhova, A.; Mehrabi, A. Markers of liver regeneration—the role of growth factors and cytokines: A systematic review. BMC Surg. 2020, 20, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, A.; Gonzalez, F.J. TNF-α regulates mouse fetal hepatic maturation induced by oncostatin M and extracellular matrices. Hepatology 2004, 40, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Tsurusaki, S.; Miyata, N.; Saijou, E.; Okochi, H.; Miyajima, A.; Tanaka, M. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 2018, 67, 296–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.M.; Lee, W.L.; Hsu, C.J.; Lu, T.T.; Wang, L.H.; Xu, G.H.; Tang, C.H. Adiponectin induces oncostatin M expression in osteoblasts through the PI3K/Akt signaling pathway. Int. J. Mol. Sci. 2015, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastuschek, J.; Poetzsch, J.; Morales-Prieto, D.M.; Schleussner, E.; Markert, U.R.; Georgiev, G. Stimulation of the JAK/STAT pathway by LIF and OSM in the human granulosa cell line COV434. J. Reprod. Immunol. 2015, 108, 48–55. [Google Scholar] [CrossRef]
- Sarkozi, R.; Hauser, C.; Noppert, S.J.; Kronbichler, A.; Pirklbauer, M.; Haller, V.M.; Grillari, J.; Grillari-Voglauer, R.; Mayer, G.; Schramek, H. Oncostatin M is a novel inhibitor of TGF-beta1-induced matricellular protein expression. Am. J. Physiol. Renal Physiol. 2011, 301, F1014–F1025. [Google Scholar] [CrossRef]
- Hamada, T.; Sato, A.; Torii, I.; Fujimoto, J.; Tsujimura, T. 541 THE role of oncostatin M in rat liver regeneration. J. Hepatol. 2008, 48, S203. [Google Scholar] [CrossRef]
- Böhmer, O.; Ehlting, C.; Hahnel, M.J.; Thomas, M.; Zanger, U.M.; Gaestel, M.; Knoefel, W.T.; Schulte am Esch, J.; Häussinger, D.; Bode, J.G. MK2 mediates induction and sensing of Oncostatin M-induced intercellular signalling in the liver but is dispensable for negative feedback regulation via SOCS3. Z. Gastroenterol. 2015, 53, A1_36. [Google Scholar] [CrossRef]
- Nakamura, K.; Nonaka, H.; Saito, H.; Tanaka, M.; Miyajima, A. Hepatocyte proliferation and tissue remodeling is impaired after liver injury in oncostatin M receptor knockout mice. Hepatology 2004, 39, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Naseem, S.; Hussain, T.; Manzoor, S. Interleukin-6: A promising cytokine to support liver regeneration and adaptive immunity in liver pathologies. Cytokine Growth Factor Rev. 2018, 39, 36–45. [Google Scholar] [CrossRef]
- Cai, X.; Li, Z.; Zhang, Q.; Qu, Y.; Xu, M.; Wan, X.; Lu, L. CXCL6-EGFR-induced Kupffer cells secrete TGF-β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J. Cell. Mol. Med. 2018, 22, 5050–5061. [Google Scholar] [CrossRef]
- Meng, F.; Francis, H.; Glaser, S.; Han, Y.; DeMorrow, S.; Stokes, A.; Staloch, D.; Venter, J.; White, M.; Ueno, Y.; et al. Role of stem cell factor and granulocyte colony-stimulating factor in remodeling during liver regeneration. Hepatology 2012, 55, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Yoshiya, S.; Shirabe, K.; Imai, D.; Toshima, T.; Yamashita, Y.I.; Ikegami, T.; Okano, S.; Yoshizumi, T.; Kawanaka, H.; Maehara, Y. Blockade of the apelin–APJ system promotes mouse liver regeneration by activating Kupffer cells after partial hepatectomy. J. Gastroenterol. 2015, 50, 573–582. [Google Scholar] [CrossRef]
- Jung, J.; Moon, J.W.; Choi, J.H.; Lee, Y.W.; Park, S.H.; Kim, G.J. Epigenetic alterations of IL-6/STAT3 signaling by placental stem cells promote hepatic regeneration in a rat model with CCl4-induced liver injury. Int. J. Stem Cells 2015, 8, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Chijiiwa, K.; Kameoka, N.; Yamaguchi, K.; Kuroki, S.; Tanaka, M. Gadolinium pretreatment decreases survival and impairs liver regeneration after partial hepatectomy under ischemia/reperfusion in rats. Surgery 2000, 127, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Austyn, J.M.; Gordon, S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 1981, 11, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Valizadeh, A.; Majidinia, M.; Samadi-Kafil, H.; Yousefi, M.; Yousefi, B. The roles of signaling pathways in liver repair and regeneration. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Oe, S.; Lemmer, E.R.; Conner, E.A.; Factor, V.M.; Leveen, P.; Larsson, J.; Karlsson, S.; Thorgeirsson, S.S. Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology 2004, 40, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, I.; Araujo, A.; Abaurrea, A.; Rezola, R.; Caffarel, M.M. Abstract P2-06-16: New targets in triple negative breast cancer: Role of Oncostatin M receptor pathway. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Vollmer, S.; Kappler, V.; Kaczor, J.; Flugel, D.; Rolvering, C.; Kato, N.; Kietzmann, T.; Behrmann, I.; Haan, C. Hypoxia-inducible factor 1alpha is up-regulated by oncostatin M and participates in oncostatin M signaling. Hepatology 2009, 50, 253–260. [Google Scholar] [CrossRef]
- Sarkozi, R.; Flucher, K.; Haller, V.; Pirklbauer, M.; Mayer, G.; Schramek, H. Oncostatin M inhibits TGF-β1-induced CTGF expression via STAT3 in human proximal tubular cells. Biochem. Biophys. Res. Commun. 2012, 424, 801–806. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| F4/80 | CTGCACCTGTAAACGAGGCTT | GCAGACTGAGTTAGGACCACAA |
| CD11b | CCATGACCTTCCAAGAGAATGC | ACCGGCTTGTGCTGTAGTC |
| GFAP | CGGAGACGCATCACCTCTG | TGGAGGAGTCATTCGAGACAA |
| Desmin | CCTGGAGCGCAGAATCGAAT | TGAGTCAAGTCTGAAACCTTGGA |
| β-actin | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
| HGF | AACAGGGGCTTTACGTTCACT | CGTCCCTTTATAGCTGCCTCC |
| TNF-α | CTGAACTTCGGGGTGATCGG | GGCTTGTCACTCGAATTTTGAGA |
| IL-6 | CTGCAAGAGACTTCCATCCAG | AGTGGTATAGACAGGTCTGTTGG |
| EGF | AGAGCATCTCTCGGATTGACC | CCCGTTAAGGAAAACTCTTAGCA |
| TGF-α | CACTCTGGGTACGTGGGTG | CACAGGTGATAATGAGGACAGC |
| TGF-β1 | CTTCAATACGTCAGACATTCGGG | GTAACGCCAGGAATTGTTGCTA |
| TGF-β2 | TCGACATGGATCAGTTTATGCG | CCCTGGTACTGTTGTAGATGGA |
| OSM | CCCGGCACAATATCCTCGG | TCTGGTGTTGTAGTGGACCGT |
| FGF1 | GGGGAGATCACAACCTTCGC | GTCCCTTGTCCCATCCACG |
| TGF-β3 | GGACTTCGGCCACATCAAGAA | TAGGGGACGTGGGTCATCAC |
| FGF2 | GCGACCCACACGTCAAACTA | CCGTCCATCTTCCTTCATAGC |
| PEDF | CCAACTTCGGCTACGATCTGT | TCTGTTCGATGTTCAGCTCCC |
| TIMP-1 | CGAGACCACCTTATACCAGCG | ATGACTGGGGTGTAGGCGTA |
| TIMP-2 | TCAGAGCCAAAGCAGTGAGC | GCCGTGTAGATAAACTCGATGTC |
| Met | GTGAACATGAAGTATCAGCTCCC | TGTAGTTTGTGGCTCCGAGAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Q.; Shen, H.; Yu, H.; Fu, J.; Dong, H.; Chen, Y.; Wang, H. F4/80+ Kupffer Cell-Derived Oncostatin M Sustains the Progression Phase of Liver Regeneration through Inhibition of TGF-β2 Pathway. Molecules 2021, 26, 2231. https://doi.org/10.3390/molecules26082231
Lu Q, Shen H, Yu H, Fu J, Dong H, Chen Y, Wang H. F4/80+ Kupffer Cell-Derived Oncostatin M Sustains the Progression Phase of Liver Regeneration through Inhibition of TGF-β2 Pathway. Molecules. 2021; 26(8):2231. https://doi.org/10.3390/molecules26082231
Chicago/Turabian StyleLu, Qingjun, Hao Shen, Han Yu, Jing Fu, Hui Dong, Yao Chen, and Hongyang Wang. 2021. "F4/80+ Kupffer Cell-Derived Oncostatin M Sustains the Progression Phase of Liver Regeneration through Inhibition of TGF-β2 Pathway" Molecules 26, no. 8: 2231. https://doi.org/10.3390/molecules26082231
APA StyleLu, Q., Shen, H., Yu, H., Fu, J., Dong, H., Chen, Y., & Wang, H. (2021). F4/80+ Kupffer Cell-Derived Oncostatin M Sustains the Progression Phase of Liver Regeneration through Inhibition of TGF-β2 Pathway. Molecules, 26(8), 2231. https://doi.org/10.3390/molecules26082231