
Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses


 , , ,
, , ,  ,
,  ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Antiviral Activity Study
2.2.1. Influenza Virus
2.2.2. Filovirus Infections
2.2.3. Hantavirus
2.2.4. Vaccinia Virus
2.3. Search for a Possible Target by Molecular Modelling Study
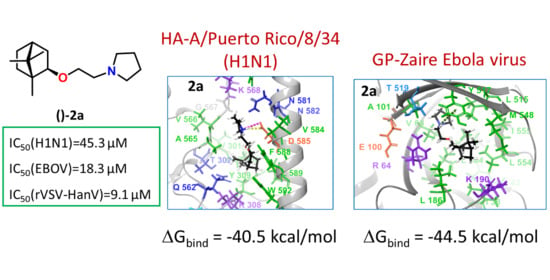
2.3.1. Binding Site Analysis
2.3.2. Molecular Modelling Study of Synthesized Derivatives to Binding Sites of HA and GP
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. General Information
4.1.2. Synthesis of 2-(2-bromoethoxy)-1,7,7-trimethylbicyclo(2.2.1)eptanes 1a and 2-(3-bromopropoxy)-1,7,7-trimethylbicyclo(2.2.1)heptanes 1b
4.1.3. General Procedure for Synthesis of Derivatives 2–8 a–b
4.2. Biological Studies
4.2.1. Evaluation of the Anti-Vaccinia Virus Activity
4.2.2. Evaluation of the Anti-Influenza Virus Activity
4.2.3. Evaluation of the Anti-Filovirus Activity
4.2.4. Evaluation of the Anti-Hantavirus Activity
4.3. Molecular Docking Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef]
- Ka-Wai Hui, E. Reasons for the increase in emerging and re-emerging viral infectious diseases. Microbes Infect. 2006, 8, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Beer, E.M.; Bhargavi Rao, V. A systematic review of the epidemiology of human monkeypox outbreaks and implications for outbreak strategy. PLoS Negl. Trop. Dis. 2019, 13, e0007791. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, M.J.M.; Jaffe, H.S.; Martin, J.C.; Stagg, R.J. Cidofovir, a new agent with potent anti-herpesvirus activity. Antivir. Chem. Chemother. 1996, 7, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Tam, R.C.; Lau, J.Y.N.; Hong, Z. Mechanisms of action of ribavirin in antiviral therapies. Antivir. Chem. Chemother. 2001, 12, 261–272. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 2014. [Google Scholar] [CrossRef]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Bormotov, N.I.; Shishkina, L.N.; Salakhutdinov, N.F. Discovery of a New Class of Inhibitors of Vaccinia Virus Based on (−)-Borneol from Abies sibirica and (+)-Camphor. Chem. Biodivers. 2018, 15. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Semenova, M.D.; Shtro, A.A.; Orshanskaya, I.R.; Zarubaev, V.V.; Salakhutdinov, N.F. Synthesis and in vitro study of novel borneol derivatives as potent inhibitors of the influenza A virus. MedChemComm 2017, 8, 960–963. [Google Scholar] [CrossRef] [Green Version]
- Kononova, A.A.; Sokolova, A.S.; Cheresiz, S.V.; Yarovaya, O.I.; Nikitina, R.A.; Chepurnov, A.A.; Pokrovsky, A.G.; Salakhutdinov, N.F. N-Heterocyclic borneol derivatives as inhibitors of Marburg virus glycoprotein-mediated VSIV pseudotype entry. MedChemComm 2017, 8, 2233–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolova, A.S.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakova, N.S.; Zaykovskaya, A.V.; Baev, D.S.; Tolstikova, T.G.; Shcherbakov, D.N.; Pyankov, O.V.; et al. Monoterpenoid-based inhibitors of filoviruses targeting the glycoprotein-mediated entry process. Eur. J. Med. Chem. 2020, 207. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Kovaleva, K.S.; Yarovaya, O.I.; Bormotov, N.I.; Shishkina, L.N.; Serova, O.A.; Sergeev, A.A.; Agafonov, A.P.; Maksuytov, R.A.; Salakhutdinov, N.F. (+)-Camphor and (−) borneol derivatives as potential anti orthopoxvirus agents. Arch. Pharm. 2021, e2100038. [Google Scholar] [CrossRef]
- Rogachev, A.D.; Putilova, V.P.; Zaykovskaya, A.V.; Yarovaya, O.I.; Sokolova, A.S.; Fomenko, V.V.; Pyankov, O.V.; Maksyutov, R.A.; Pokrovsky, A.G. Biostability study, quantitation method and preliminary pharmacokinetics of a new antifilovirus agent based on borneol and 3-(piperidin-1-yl)propanoic acid. J. Pharm. Biomed. Anal. 2021, 114062. [Google Scholar] [CrossRef]
- Snape, T.J.; Astles, A.M.; Davies, J. Understanding the chemical basis of drug stability and degradation. Pharm. J. 2010, 285, 416–417. [Google Scholar]
- Fedorova, I.V.; Chukicheva, I.Y.; Shumova, O.A.; Kutchin, A.V. Synthesis of phenolic antioxidants with isobornyl and tert-butyl fragments. Russ. J. Gen. Chem. 2013, 83, 1103–1110. [Google Scholar] [CrossRef]
- Ren, J.; Zhao, Y.; Fry, E.E.; Stuart, D.I. Target Identification and Mode of Action of Four Chemically Divergent Drugs against Ebolavirus Infection. J. Med. Chem. 2018, 61, 724–733. [Google Scholar] [CrossRef]
- Johansen, L.M.; DeWald, L.E.; Shoemaker, C.J.; Hoffstrom, B.G.; Lear-Rooney, C.M.; Stossel, A.; Nelson, E.; Delos, S.E.; Simmons, J.A.; Grenier, J.M.; et al. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Manigold, T.; Vial, P. Human hantavirus infections: Epidemiology, clinical features, pathogenesis and immunology. Swiss Med. Wkly. 2014, 144, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bi, Z.; Formenty, P.B.H.; Roth, C.E. Hantavirus infection: A review and global update. J. Infect. Dev. Ctries. 2008, 2, 3–23. [Google Scholar] [CrossRef]
- Byrd, C.M.; Bolken, T.C.; Mjalli, A.M.; Arimilli, M.N.; Andrews, R.C.; Rothlein, R.; Andrea, T.; Rao, M.; Owens, K.L.; Hruby, D.E. New Class of Orthopoxvirus Antiviral Drugs That Block Viral Maturation. J. Virol. 2004, 78, 12147–12156. [Google Scholar] [CrossRef] [Green Version]
- Selivanov, B.A.; Tikhonov, A.Y.; Belanov, E.F.; Bormotov, N.I.; Kabanov, A.S.; Mazurkov, O.Y.; Serova, O.A.; Shishkina, L.N.; Agafonov, A.P.; Sergeev, A.N. Synthesis and Antiviral Activity of 1-Aryl-3-(3,5-Dioxo-4-Azatetracyclo-[5.3.2.02,6.08,10]Dodec-11-EN-4-YL)Ureas. Pharm. Chem. J. 2017, 51, 439–443. [Google Scholar] [CrossRef]
- Pattnaik, G.P.; Chakraborty, H. Entry Inhibitors: Efficient Means to Block Viral Infection. J. Membr. Biol. 2020, 253, 425–444. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisevich, S.S.; Gureev, M.A.; Yarovaya, O.I.; Zarubaev, V.V.; Kostin, G.A.; Porozov, Y.B.; Salakhutdinov, N.F. Can molecular dynamics explain decreased pathogenicity in mutant camphecene-resistant influenza virus? J. Biomol. Struct. Dyn. 2021. [Google Scholar] [CrossRef] [PubMed]
- Leiva, R.; Barniol-Xicota, M.; Codony, S.; Ginex, T.; Vanderlinden, E.; Montes, M.; Caffrey, M.; Luque, F.J.; Naesens, L.; Vázquez, S. Aniline-Based Inhibitors of Influenza H1N1 Virus Acting on Hemagglutinin-Mediated Fusion. J. Med. Chem. 2018, 61, 98–118. [Google Scholar] [CrossRef]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Li, R.; Zhou, Y.; Xiao, M.; Ma, C.; Yang, Z.; Zeng, S.; Du, Q.; Yang, C.; Jiang, H.; et al. Discovery of Highly Potent Pinanamine-Based Inhibitors against Amantadine- and Oseltamivir-Resistant Influenza A Viruses. J. Med. Chem. 2018, 61, 5187–5198. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, K.S.; Zubkov, F.I.; Bormotov, N.I.; Novikov, R.A.; Dorovatovskii, P.V.; Khrustalev, V.N.; Gatilov, Y.V.; Zarubaev, V.V.; Yarovaya, O.I.; Shishkina, L.N.; et al. Synthesis of d-(+)-camphor-based: N -acylhydrazones and their antiviral activity. Medchemcomm 2018, 9, 2072–2082. [Google Scholar] [CrossRef]
- Whitt, M.A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 2010, 169, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Melorose, J.; Perroy, R.; Careas, S. Methods in Molecular Biology Polyamine Protocols; Humana Press: Totowa, NJ, USA, 2015; Volume 1, ISBN 9788578110796. [Google Scholar]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The Structure and Receptor Binding Properties of the 1918 Influenza Hemagglutinin. Science 2004, 303, 1838–1842. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Watson, M.A.; Greenwood, J.R.; Philipp, D.M. Multiconformation, Density Functional Theory-Based pKa Prediction in Application to Large, Flexible Organic Molecules with Diverse Functional Groups. J. Chem. Theory Comput. 2016, 12, 6001–6019. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the prediction of absolute solvation free energies using the next generation opls force field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CC50 (µM) a | IC50 H1N1 (µM) b | SI c |
|---|---|---|---|
| 2a | >1193.3 | 45.3 ± 5.2 | 26 |
| 2b | 71.9 ± 4.4 | 16.8 ± 2.3 | 4 |
| 3a | >1130.2 | 64.8 ± 5.9 | 17 |
| 3b | 949.9 ± 52.5 | 346.7 ± 41.1 | 2.5 |
| 4a | >1073.5 | 37.9 ± 5.6 | 28 |
| 4b | 491.0 ± 32.2 | 69.7 ± 8.4 | 7 |
| 5a | 28.5 ± 1.2 | 13.2 ± 2.0 | 2 |
| 5b | 51.0 ± 4.1 | >37.4 | 1 |
| 6a | 33.7 ± 2.8 | 3.4 ± 0.5 | 10 |
| 6b | 10.8 ± 0.8 | >8 | 1 |
| 7a | 385.2 ± 19.7 | 252.0 ± 31.1 | 1 |
| 7b | 561.8 ± 48.2 | 24.2 ± 3.1 | 23 |
| 8a | 50.5 ± 4.5 | 35.3 ± 5.0 | 1 |
| 8b | 374.2 ± 22.8 | 320.0 ± 36.9 | 1 |
| Ribavirin | >2000 | 24.6 ± 3.7 | >81 |
| Rimantadine | 335 ± 27 | 67.0 ± 4.9 | 5 |
| Compound | CC50 (µM) a | IC50 (µM) | SIEboV-GP e | ||
|---|---|---|---|---|---|
| EboV-GP b | MarV-GP c | VSV-G d | |||
| 2a | 497.2 ± 8.0 | 0.12 ± 0.04 | 73.6 ± 1.9 | 123.3 ± 4.0 | 4166 |
| 2b | 107.9 ± 4.0 | NA | 63.9 ± 2.0 | NT | - |
| 3a | 941.9 ± 18.8 | 6.3 ±0.3 | NA | 941.9 ± 18.8 | 150 |
| 3b | 2119.3 ± 49.2 | 473.1 ± 11.4 | 170.3 ± 3.8 | 946.1 ± 22.7 | 4 |
| 4a | 447.3 ± 10.7 | 1.3 ± 0.1 | 60.8 ± 1.8 | 110.9 ± 3.6 | 357 |
| 4b | 140.8 ± 3.4 | NA | 96.1 ± 3.4 | NT | - |
| 5a | 652.9 ± 14.3 | NA | 85.6 ± 1.8 | NT | - |
| 5b | 108.7 ± 3.4 | NA | NA | NT | - |
| 6a | 575.4 ± 11.2 | NA | NA | NT | - |
| 6b | 148.5 ± 5.4 | 67.5 ± 2.7 | 124.2 ± 2.7 | NT | 2 |
| 7a | 860.1 ± 22.4 | 0.6 ± 0.2 | 149.6 ± 3.7 | 934.9 ± 26.2 | 1433 |
| 7b | 1440.1 ± 28.4 | NA | NA | NA | - |
| 8a | 721.4 ± 14.4 | 112.5 ± 3.6 | 90.2 ± 3.6 | 360.7 ± 10.8 | 6 |
| 8b | 703.8 ± 10.3 | NA | NA | NT | - |
| Sertraline | 408 ± 35.9 | 0.7 ± 0.07 | NT | 582 | |
| Compound | CC50 (µM) a | IC50 EBOV (µM) b | SI EBOV c |
|---|---|---|---|
| 2a | 230.7 ± 16.5 | 18.3 ± 3.1 | 12 |
| 3a | 55.9 ± 12.2 | 15.2 ± 2.1 | 3.6 |
| 4a | 40.2 ± 6.9 | NA | - |
| 7a | 57.5 ± 13.5 | 5.6 ± 0.9 | 10 |
| Sertraline d | 3.7 ± 0.6 |
| Compound | CC50 (µM) a | IC50 VV (µM) b | Compound | CC50 (µM) a | IC50 VV (µM) b |
|---|---|---|---|---|---|
| 2b | 62.3 ± 3.6 | NA | 6b | 21.3 ± 2.2 | NA |
| 3b | 139.6 ± 8.7 | NA | 7a | 171.6 ± 10.8 | NA |
| 4a | 11.1 ± 1.4 | NA | 7b | 173.5 ± 11.9 | NA |
| 4b | 40.9 ± 2.7 | NA | 8a | 19.1 ± 2.5 | NA |
| 5a | 100.3 ± 6.1 | NA | 8b | 23.7 ± 2.7 | NA |
| 5b | 64.2 ± 3.1 | NA | Cidofovir | 475.3 ± 30.1 | 40.0 ± 1.2 |
| 6a | 14.0 ± 1.7 | NA | ST-246 | 1276 ± 202 | 0.01 ± 0.003 |
| Compound | CC50 (µM) a | IC50 rVSV-ΔG-Gn-Gc (µM) b | SI c |
|---|---|---|---|
| 2a | 358.0 ± 27.8 | 9.1 ± 1.2 | 39 |
| 3a | 150.7 ± 18.8 | 5.0 ± 0.8 | 30 |
| 7a | 1159.3 ± 56.1 | 14.8 ± 1.9 | 78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokolova, A.S.; Putilova, V.P.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; Shcherbakov, D.N.; Orshanskaya, I.R.; Sinegubova, E.O.; Esaulkova, I.L.; et al. Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses. Molecules 2021, 26, 2235. https://doi.org/10.3390/molecules26082235
Sokolova AS, Putilova VP, Yarovaya OI, Zybkina AV, Mordvinova ED, Zaykovskaya AV, Shcherbakov DN, Orshanskaya IR, Sinegubova EO, Esaulkova IL, et al. Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses. Molecules. 2021; 26(8):2235. https://doi.org/10.3390/molecules26082235
Chicago/Turabian StyleSokolova, Anastasiya S., Valentina P. Putilova, Olga I. Yarovaya, Anastasiya V. Zybkina, Ekaterina D. Mordvinova, Anna V. Zaykovskaya, Dmitriy N. Shcherbakov, Iana R. Orshanskaya, Ekaterina O. Sinegubova, Iana L. Esaulkova, and et al. 2021. "Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses" Molecules 26, no. 8: 2235. https://doi.org/10.3390/molecules26082235
APA StyleSokolova, A. S., Putilova, V. P., Yarovaya, O. I., Zybkina, A. V., Mordvinova, E. D., Zaykovskaya, A. V., Shcherbakov, D. N., Orshanskaya, I. R., Sinegubova, E. O., Esaulkova, I. L., Borisevich, S. S., Bormotov, N. I., Shishkina, L. N., Zarubaev, V. V., Pyankov, O. V., Maksyutov, R. A., & Salakhutdinov, N. F. (2021). Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses. Molecules, 26(8), 2235. https://doi.org/10.3390/molecules26082235