
The Interaction of Human Glutathione Transferase GSTA1-1 with Reactive Dyes

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
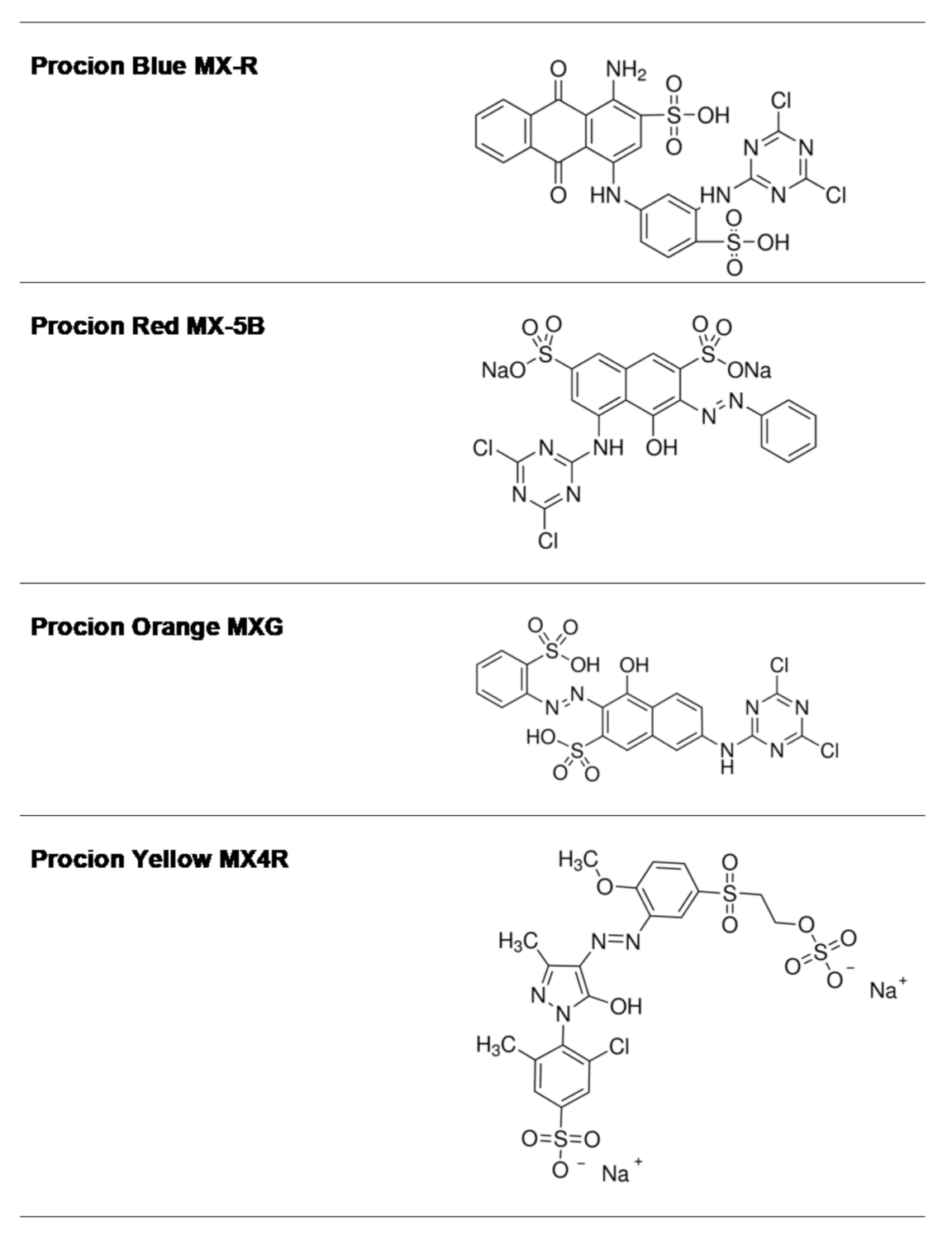
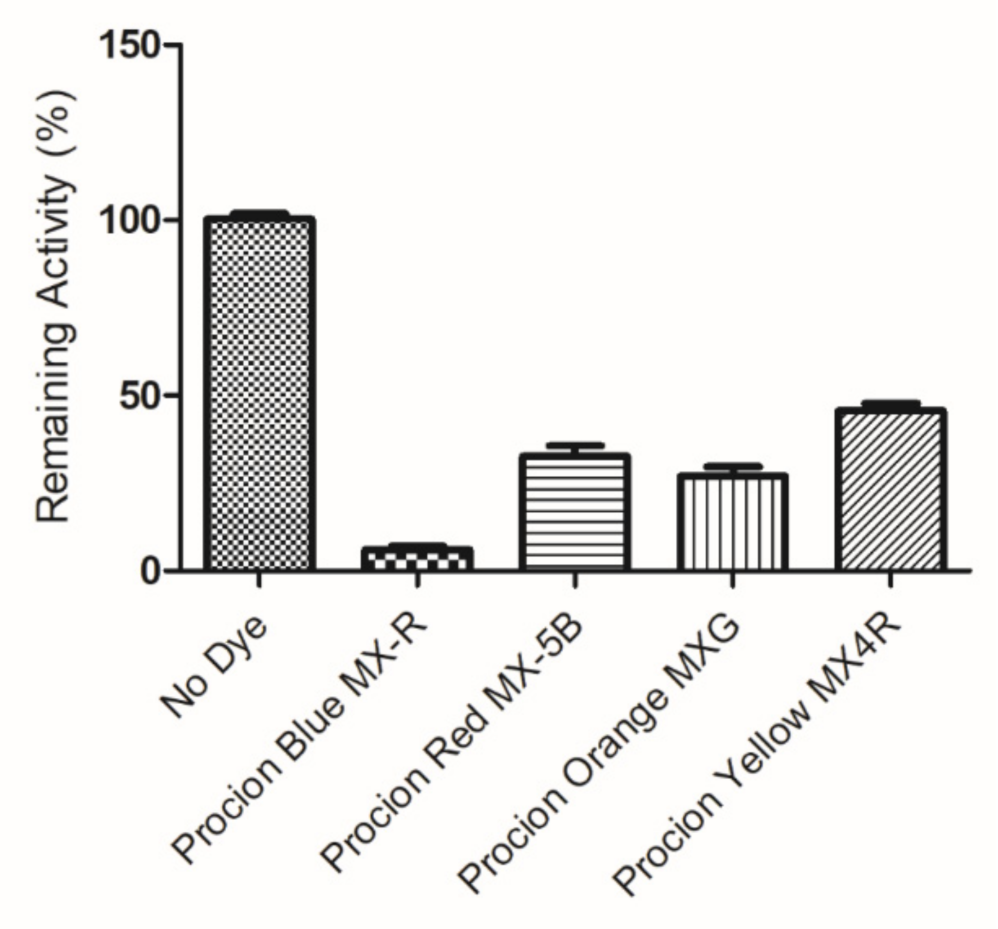
2.1. Screening of the Inhibition Potency of DCTD
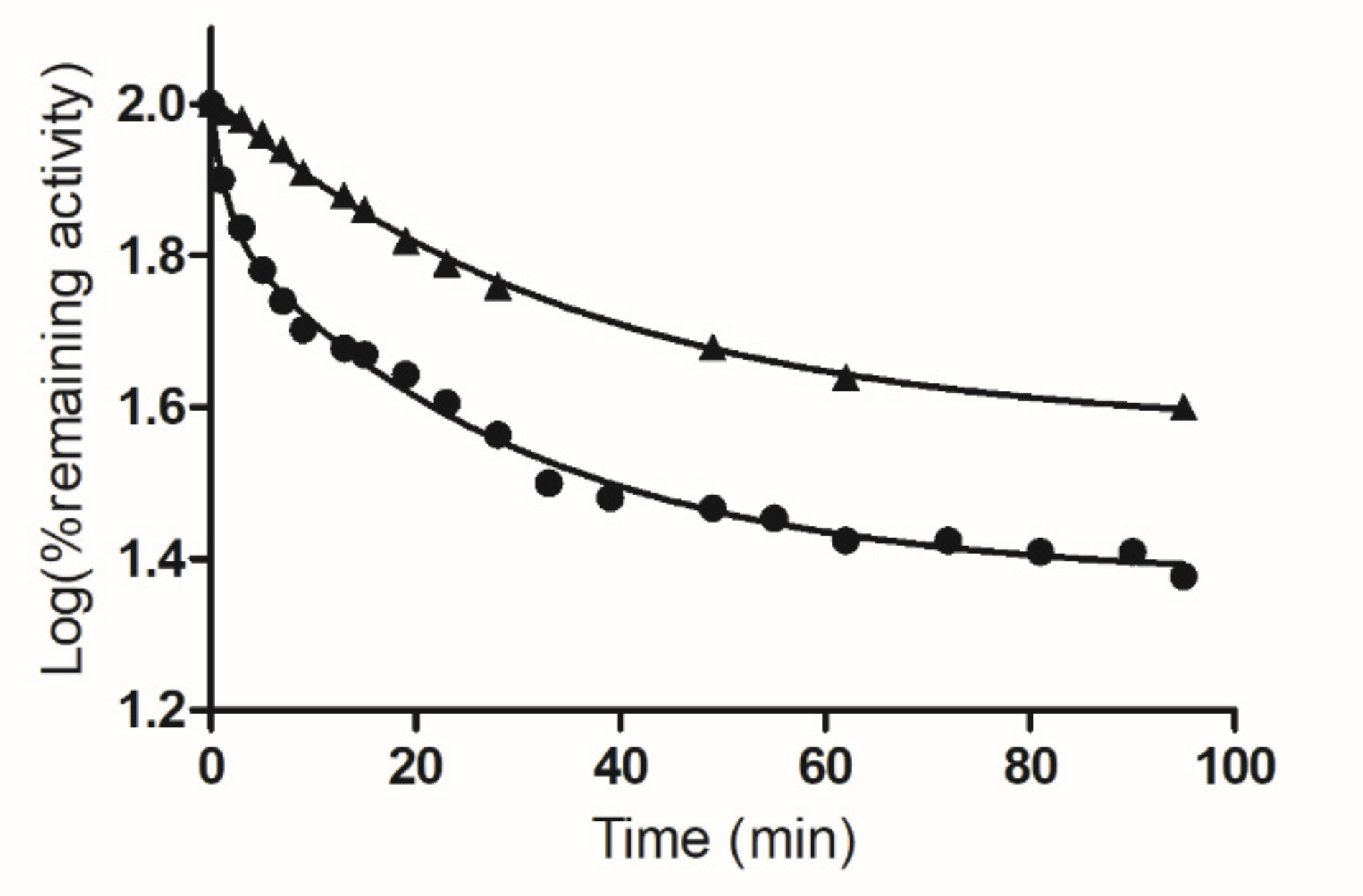
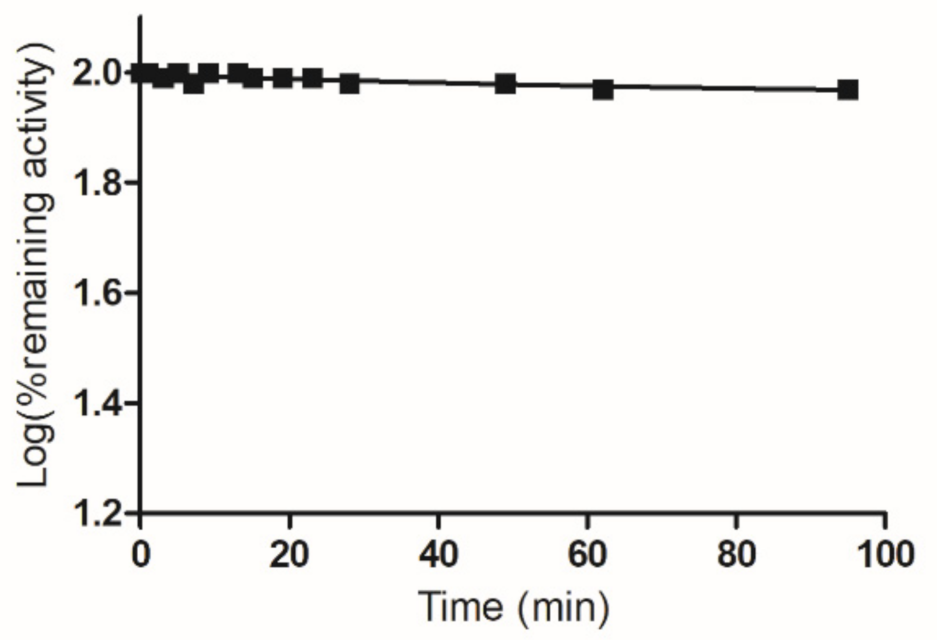
2.2. Inactivation of hGSTA1-1 by PBMX-R
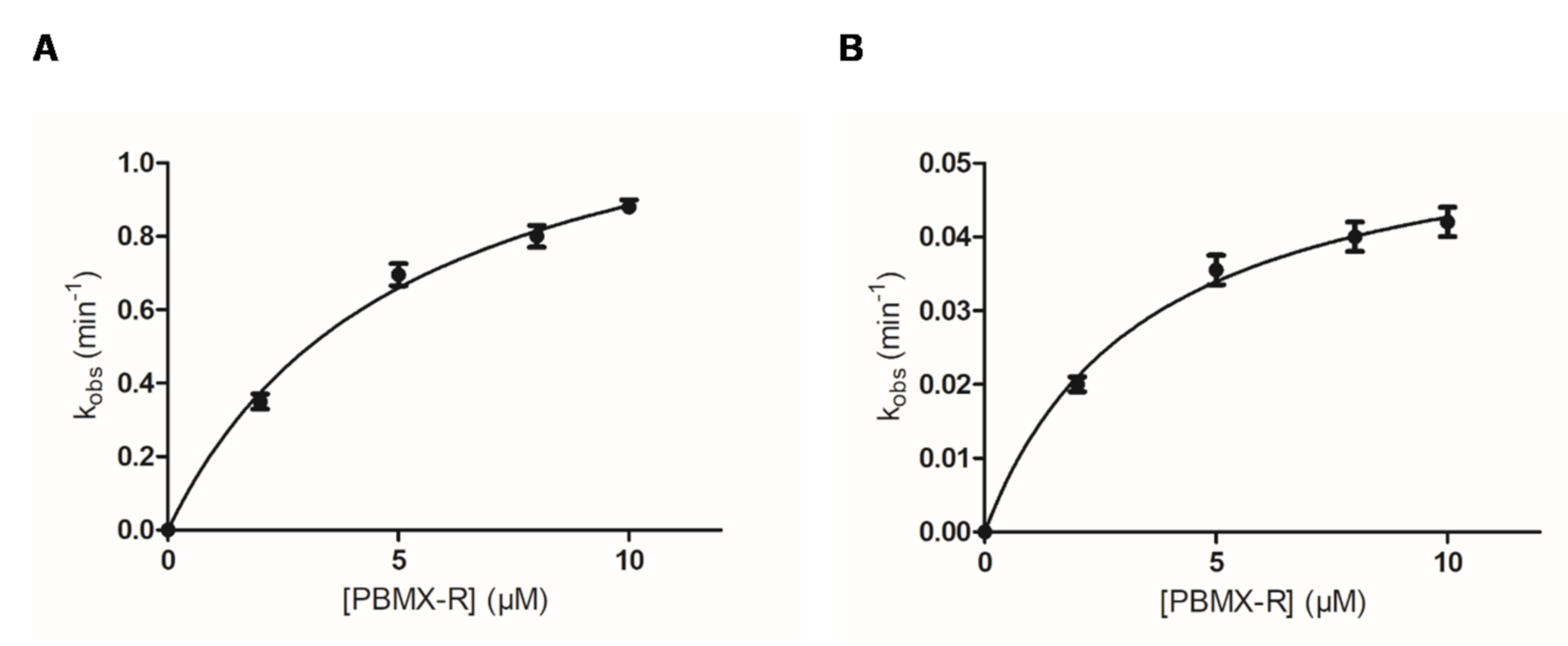
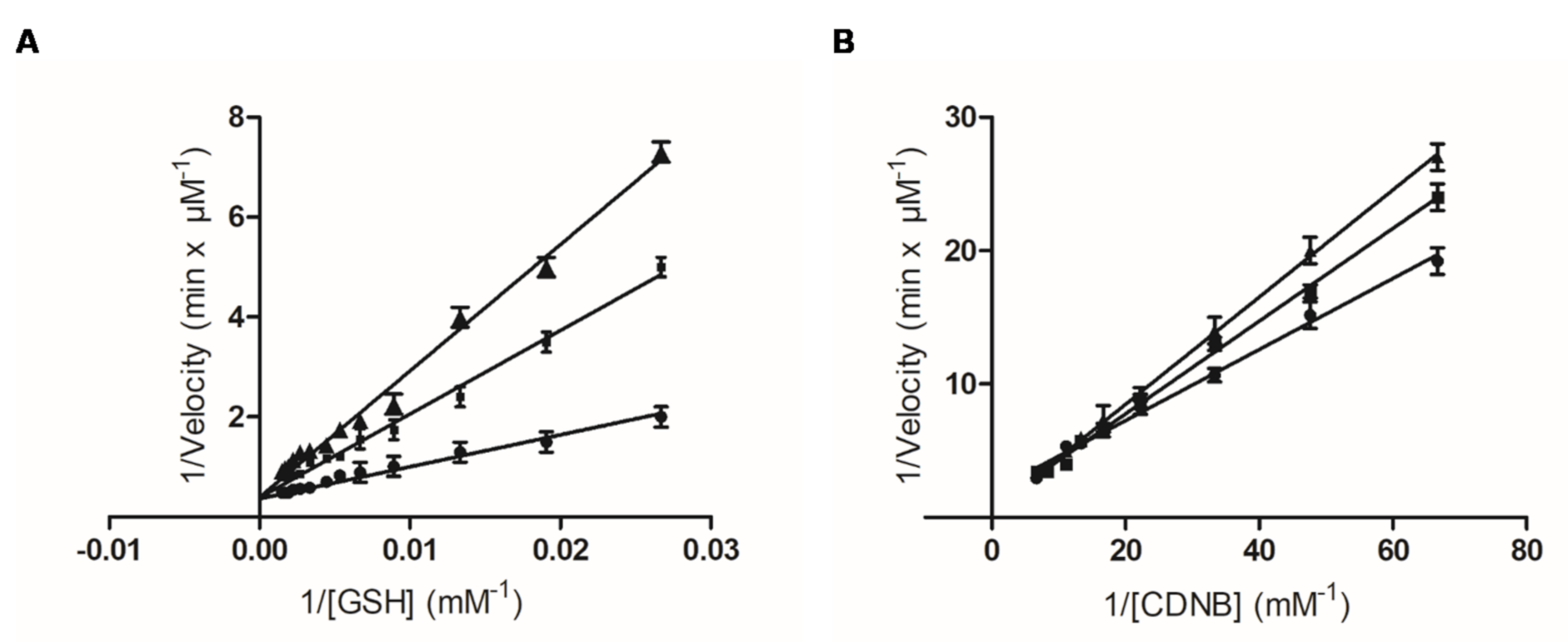
2.3. Kinetic Inhibition Analysis
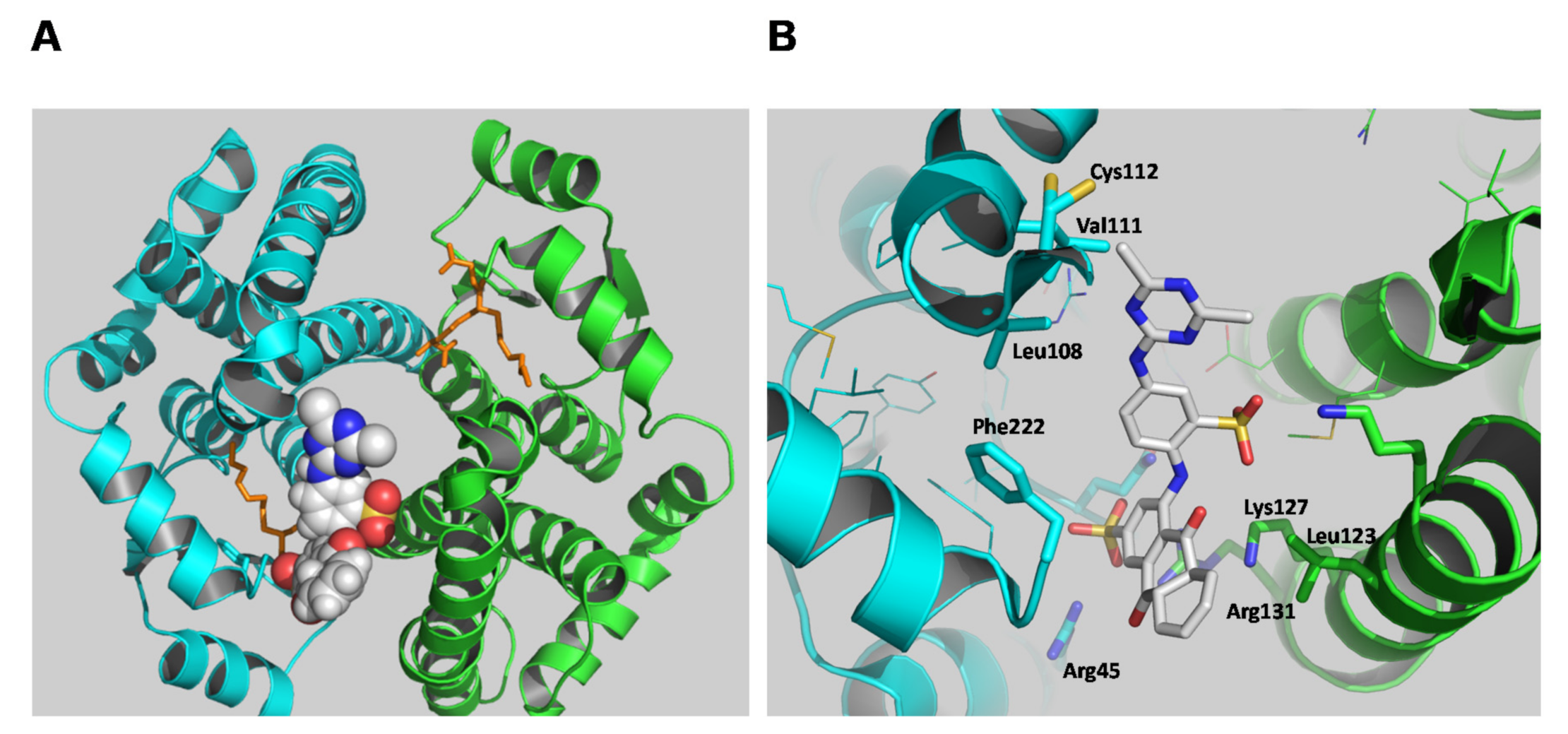
2.4. The Identification of hGSTA1-1 Residue Was Modified by PBMX-R
3. Materials and Methods
3.1. Materials
3.2. Heterologous Expression and Purification of Recombinant Human Glutathione Transferase A1-1
3.3. Assay of GST Activity and Inhibition Analysis by Triazine Dyes
3.4. Enzyme Inactivation Studies by PBMX-R
3.5. Modification of hGSTA1-1 with DTNB and N-ethylmaleimide
3.6. The Interaction of PBMX-R with hGSTA1-1 by Molecular Docking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CB3GA | Cibacron blue 3GA |
| CDNB | 1-chloro-2,4-dinitrobenzene |
| DCTD | dichlorotriazine dye |
| GSH | glutathione |
| GST | glutathione transferase |
| G-site | glutathione-binding site |
| hGSTA1-1 | human glutathione transferase A1-1 |
| hPBMX-R | hydrolyzed PBMX-R |
| H-ste | hydrophobic binding site for electrophilic substrates |
| PBMX-R | Procion blue MX-R |
| L-site | ligandin-binding site; (L-site) |
| S-pNb-GSH | S-p-nitrobenzyl-glutathione |
References
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Sinning, I.; Kleywegt, G.J.; Cowan, S.W.; Reinemer, P.; Dirr, H.W.; Huber, R.; Gilliland, G.L.; Armstrong, R.N.; Ji, X.; Board, P.G.; et al. Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J. Mol. Biol. 1993, 232, 192–212. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B. The isoenzymes of glutathione transferase. Advan. Enzymol. Rel. Areas Mol. Biol. 1985, 57, 357–417. [Google Scholar]
- Mannervik, B. Five decades with glutathione and the GSTome. J. Biol. Chem. 2012, 287, 6072–6083. [Google Scholar] [CrossRef] [Green Version]
- Perperopoulou, F.; Pouliou, F.; Labrou, N.E. Recent advances in protein engineering and biotechnological applications of glutathione transferases. Crit. Rev. Biotechnol. 2018, 38, 511–528. [Google Scholar] [CrossRef]
- Mohana, K.; Achary, A. Human cytosolic glutathione-S-transferases: Quantitative analysis of expression, comparative analysis of structures and inhibition strategies of isozymes involved in drug resistance. Drug Metab. Rev. 2017, 49, 318–337. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione Transferases: Potential Targets to Overcome Chemoresistance in Solid Tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef] [Green Version]
- Karpusas, M.; Axarli, I.; Chiniadis, L.; Papakyriakou, A.; Bethanis, K.; Scopelitou, K.; Clonis, Y.D.; Labrou, N.E. The interaction of the chemotherapeutic drug chlorambucil with human glutathione transferase A1-1: Kinetic and structural analysis. PLoS ONE 2013, 8, e56337. [Google Scholar] [CrossRef] [Green Version]
- Smitherman, P.K.; Townsend, A.J.; Kute, T.E.; Morrow, C.S. Role of multidrug resistance protein 2 (MRP2, ABCC2) in alkylating agent detoxification: MRP2 potentiates glutathione S-transferase A1-1-mediated resistance to chlorambucil cytotoxicity. J. Pharmacol. Exp. Ther. 2004, 308, 260–267. [Google Scholar] [CrossRef]
- Sharma, R.; Ellis, B.; Sharma, A. Role of alpha class glutathione transferases (GSTs) in chemoprevention: GSTA1 and A4 overexpressing human leukemia (HL60) cells resist sulforaphane and curcumin induced toxicity. Phytother. Res. 2011, 25, 563–568. [Google Scholar] [CrossRef]
- Ricci, G.; De Maria, F.; Antonini, G.; Turella, P.; Bullo, A.; Stella, L.; Filomeni, G.; Federici, G.; Caccuri, A.M. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J. Biol. Chem. 2005, 280, 26397–26405. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Hu, X.; Xu, B.; Tong, T.; Jing, Y.; Xi, L.; Zhou, W.; Lu, J.; Wang, X.; Yang, X.; et al. Glutathione S transferase isozyme alpha 1 is predominantly involved in the cisplatin resistance of common types of solid cancer. Oncol. Rep. 2019, 41, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Martin, A.; Nirgude, S.; Chaudhary, B.; Mondal, S.; Sarkar, A. Quinacrine inhibits GSTA1 activity and induces apoptosis through G1/S arrest and generation of ROS in human non-small cell lung cancer cell lines. Oncotarget 2020, 11, 1603–1617, Erratum in: Oncotarget 2020, 11, 4364–4365. [Google Scholar] [CrossRef]
- Guo, W.; Tan, H.Y.; Chen, F.; Wang, N.; Feng, Y. Targeting Cancer Metabolism to Resensitize Chemotherapy: Potential Development of Cancer Chemosensitizers from Traditional Chinese Medicines. Cancers 2020, 12, 404. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Parker, L.J.; Ang, W.H.; Rodolfo, C.; Gabbarini, V.; Hancock, N.C.; Palone, F.; Mazzetti, A.P.; Menin, L.; Morton, C.J.; et al. A structure-based mechanism of cisplatin resistance mediated by glutathione transferase P1-1. Proc. Natl. Acad. Sci. USA 2019, 116, 13943–13951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.C.; Sha, H.H.; Xu, X.Y.; Hu, T.M.; Lou, R.; Li, H.; Wu, J.Z.; Dan, C.; Feng, J. Glutathione S-transferase π: A potential role in antitumor therapy. Drug Des. Dev. Ther. 2018, 12, 3535–3547. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef]
- Ansari, M.I.; Khan, M.M.; Saquib, M.; Khatoon, S.; Hussain, M.K. Dithiolethiones: A privileged pharmacophore for anticancer therapy and chemoprevention. Future Med. Chem. 2018, 10, 1241–1260. [Google Scholar] [CrossRef]
- Sharma, A.; Patrick, B.; Li, J.; Sharma, R.; Jeyabal, P.V.; Reddy, P.M.; Awasthi, S.; Awasthi, Y.C. Glutathione S-transferases as antioxidant enzymes: Small cell lung cancer (H69) cells transfected with hGSTA1 resist doxorubicin-induced apoptosis. Arch. Biochem. Biophys. 2006, 452, 165–173. [Google Scholar] [CrossRef]
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug metabolizing enzymes and their inhibitors’ role in cancer resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef]
- Xie, Y.; Dahlin, J.L.; Oakley, A.J.; Casarotto, M.G.; Board, P.G.; Baell, J.B. Reviewing Hit Discovery Literature for Difficult Targets: Glutathione Transferase Omega-1 as an Example. J. Med. Chem. 2018, 61, 7448–7470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Elfarra, A.A. Toxicity mechanism-based prodrugs: Glutathione-dependent bioactivation as a strategy for anticancer prodrug design. Expert Opin. Drug Discov. 2018, 13, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Pouliou, F.M.; Thireou, T.N.; Eliopoulos, E.E.; Tsoungas, P.G.; Labrou, N.E.; Clonis, Y.D. Isoenzyme- and allozyme-specific inhibitors: 2,2′-dihydroxybenzophenones and their carbonyl N-analogues that discriminate between human glutathione transferase A1-1 and P1-1 allozymes. Chem. Biol. Drug Des. 2015, 86, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premetis, G.; Marugas, P.; Fanos, G.; Vlachakis, D.; Chronopoulou, E.G.; Perperopoulou, F.; Dubey, K.K.; Shukla, P.; Foudah, A.I.; Muharram, M.M.; et al. The Interaction of the Microtubule Targeting Anticancer Drug Colchicine with Human Glutathione Transferases. Curr. Pharm. Des. 2020, 26, 5205–5212. [Google Scholar] [CrossRef]
- Dirr, H.W.; Wallace, L.A. Role of the C-terminal helix 9 in the stability and ligandin function of class alpha glutathione transferase A1-1. Biochemistry 1999, 38, 15631–15640. [Google Scholar] [CrossRef]
- Grahn, E.; Novotny, M.; Jakobsson, E.; Gustafsson, A.; Grehn, L.; Olin, B.; Madsen, D.; Wahlberg, M.; Mannervik, B.; Kleywegt, G.J. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Cryst. 2006, D62, 197–207. [Google Scholar] [CrossRef]
- Misquitta, S.A.; Colman, R.F. Communication between the two active sites of glutathione S-transferase A1-1, probed using wild-type-mutant heterodimers. Biochemistry 2005, 44, 8608–8619. [Google Scholar] [CrossRef]
- Sayed, Y.; Hornby, J.A.; Lopez, M.; Dirr, H. Thermodynamics of the ligandin function of human class Alpha glutathione transferase A1-1: Energetics of organic anion ligand binding. Biochem. J. 2002, 363, 341–346. [Google Scholar] [CrossRef]
- Sayed, Y.; Wallance, L.A.; Dirr, H.W. The hydrophobic lock-and-key intersubunit motif of glutathione transferase A1-1: Implications for catalysis, ligandin function and stability. FEBS Lett. 2000, 465, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Labrou, N.E.; Clonis, Y.D. The interaction of Candida boidinii formate dehydrogenase with a new family of chimeric biomimetic dye-ligands. Arch. Biochem. Biophys. 1995, 316, 169–178. [Google Scholar] [CrossRef]
- Platis, M.; Vlachakis, D.; Foudah, A.I.; Muharram, M.M.; Alqarni, M.H.; Papageorgiou, A.C.; Labrou, N.E. The interaction of Schistosoma japonicum glutathione transferase with Cibacron blue 3GA and its fragments. Med. Chem. 2021, in press.. [Google Scholar] [CrossRef]
- Axarli, I.A.; Rigden, D.J.; Labrou, N.E. Characterization of the ligandin site of maize glutathione S-transferase I. Biochem. J. 2004, 382, 885–893. [Google Scholar] [CrossRef]
- Platis, D.; Smith, B.J.; Huyton, T.; Labrou, N.E. Structure-guided design of a novel class of benzyl-sulfonate inhibitors for influenza virus neuraminidase. Biochem. J. 2006, 399, 215–223. [Google Scholar] [CrossRef]
- Oakley, A.J.; Lo Bello, M.; Nuccetelli, M.; Mazzetti, A.P.; Parker, M.W. The ligandin (non-substrate) binding site of human Pi class glutathione transferase is located in the electrophile binding site (H-site). J. Mol. Biol. 1999, 291, 913–926. [Google Scholar] [CrossRef]
- Mosialou, E.; Morgenstern, R. Inhibition studies on rat liver microsomal glutathione transferase. Chem. Biol. Interact. 1990, 74, 275–280. [Google Scholar] [CrossRef]
- Lyon, R.P.; Atkins, W.M. Kinetic characterization of native and cysteine 112-modified glutathione S-transferase A1-1: Reassessment of nonsubstrate ligand binding. Biochemistry 2002, 41, 10920–10927. [Google Scholar] [CrossRef]
- Kumari, V.; Dyba, M.A.; Holland, R.J.; Liang, Y.H.; Singh, S.V.; Ji, X. Irreversible Inhibition of Glutathione S-Transferase by Phenethyl Isothiocyanate (PEITC), a Dietary Cancer Chemopreventive Phytochemical. PLoS ONE 2016, 11, e0163821. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Wallace, L.; Burke, J.; Stevens, J.; Dirr, H. Aflatoxin B1 and sulphobromophthalein binding to the dimeric human glutathione S-transferase A1-1: A fluorescence spectroscopic analysis. Eur. J. Biochem. 1998, 257, 434–442. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Farrotti, A.; Stella, L.; Ketterman, A.J.; Pedersen, J.Z.; Allocati, N.; Lau, P.C.; Grosse, S.; Eltis, L.D.; et al. The impact of nitric oxide toxicity on the evolution of the glutathione transferase superfamily: A proposal for an evolutionary driving force. J. Biol. Chem. 2013, 288, 24936–24947. [Google Scholar] [CrossRef] [Green Version]
- Bocedi, A.; Fabrini, R.; Lo Bello, M.; Caccuri, A.M.; Federici, G.; Mannervik, B.; Cornish-Bowden, A.; Ricci, G. Evolution of Negative Cooperativity in Glutathione Transferase Enabled Preservation of Enzyme Function. J. Biol. Chem. 2016, 291, 26739–26749. [Google Scholar] [CrossRef] [Green Version]
- Axarli, I.; Muleta, A.W.; Chronopoulou, E.G.; Papageorgiou, A.C.; Labrou, N.E. Directed evolution of glutathione transferases towards a selective glutathione-binding site and improved oxidative stability. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3416–3428. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Axarli, I.; Labrou, N.E.; Petrou, C.; Rassias, N.; Cordopatis, P.; Clonis, Y.D. Sulphonamide-based bombesin prodrug analogues for glutathione transferase, useful in targeted cancer chemotherapy. Eur. J. Med. Chem. 2009, 44, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- King, M.M.; Colman, R. Affinity labeling of nicotinamide adenine dinucleotide dependent isocitrate dehydrogenase by the 2′,3′-dialdehyde derivative of adenosine 5′-diphosphate. Evidence for the formation of an unusual reaction product. Biochemistry 1983, 22, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Kitz, R.; Wilson, I.B. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962, 237, 3245–3249. [Google Scholar] [CrossRef]
- Rendina, A.R.; Cheng, D. Characterization of the inactivation of rat fatty acid synthase by C75: Inhibition of partial reactions and protection by substrates. Biochem. J. 2005, 388, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Eyer, P.; Worek, F.; Kiderlen, D.; Sinko, G.; Stuglin, A.; Simeon-Rudolf, V.; Reiner, E. Molar absorption coefficients for the reduced Ellman reagent: Reassessment. Anal. Biochem. 2003, 312, 224–227. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2001, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- Perperopoulou, F.; Ataya, F.S.; Fouad, D.; Malik, A.; Saeed, H.M.; Labrou, N.E. Biochemical Characterization of the Detoxifying Enzyme Glutathione Transferase P1-1 from the Camel Camelus Dromedarius. Cell Biochem. Biophys. 2016, 74, 459–472. [Google Scholar] [CrossRef]
- Le Trong, I.; Stenkamp, R.E.; Ibarra, C.; Atkins, W.M.; Adman, E.T. 1.3-A resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site. Proteins 2002, 48, 618–627. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins 2007, 67, 1010–1025. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Fast | Slow |
|---|---|---|
| KD | 5.2 ± 0.5 | 3.4 ± 0.2 |
| k3 | 1.3 ± 0.1 | 0.06 ± 0.003 |
Sample Availability: More computational data are available upon request. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Labrou, N.E. The Interaction of Human Glutathione Transferase GSTA1-1 with Reactive Dyes. Molecules 2021, 26, 2399. https://doi.org/10.3390/molecules26082399
Alqarni MH, Foudah AI, Muharram MM, Labrou NE. The Interaction of Human Glutathione Transferase GSTA1-1 with Reactive Dyes. Molecules. 2021; 26(8):2399. https://doi.org/10.3390/molecules26082399
Chicago/Turabian StyleAlqarni, Mohammed Hamed, Ahmed Ibrahim Foudah, Magdy Mohamed Muharram, and Nikolaos E. Labrou. 2021. "The Interaction of Human Glutathione Transferase GSTA1-1 with Reactive Dyes" Molecules 26, no. 8: 2399. https://doi.org/10.3390/molecules26082399
APA StyleAlqarni, M. H., Foudah, A. I., Muharram, M. M., & Labrou, N. E. (2021). The Interaction of Human Glutathione Transferase GSTA1-1 with Reactive Dyes. Molecules, 26(8), 2399. https://doi.org/10.3390/molecules26082399