
Determination of Anthelmintic and Antiprotozoal Drug Residues in Fish Using Liquid Chromatography-Tandem Mass Spectrometry

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of Liquid Chromatography-Mass Spectrometry
2.2. Optimization of Sample Preparation Methods
2.2.1. Extraction
2.2.2. Clean-Up Procedure
2.3. Method Validation
2.3.1. Validation of Analytical Method
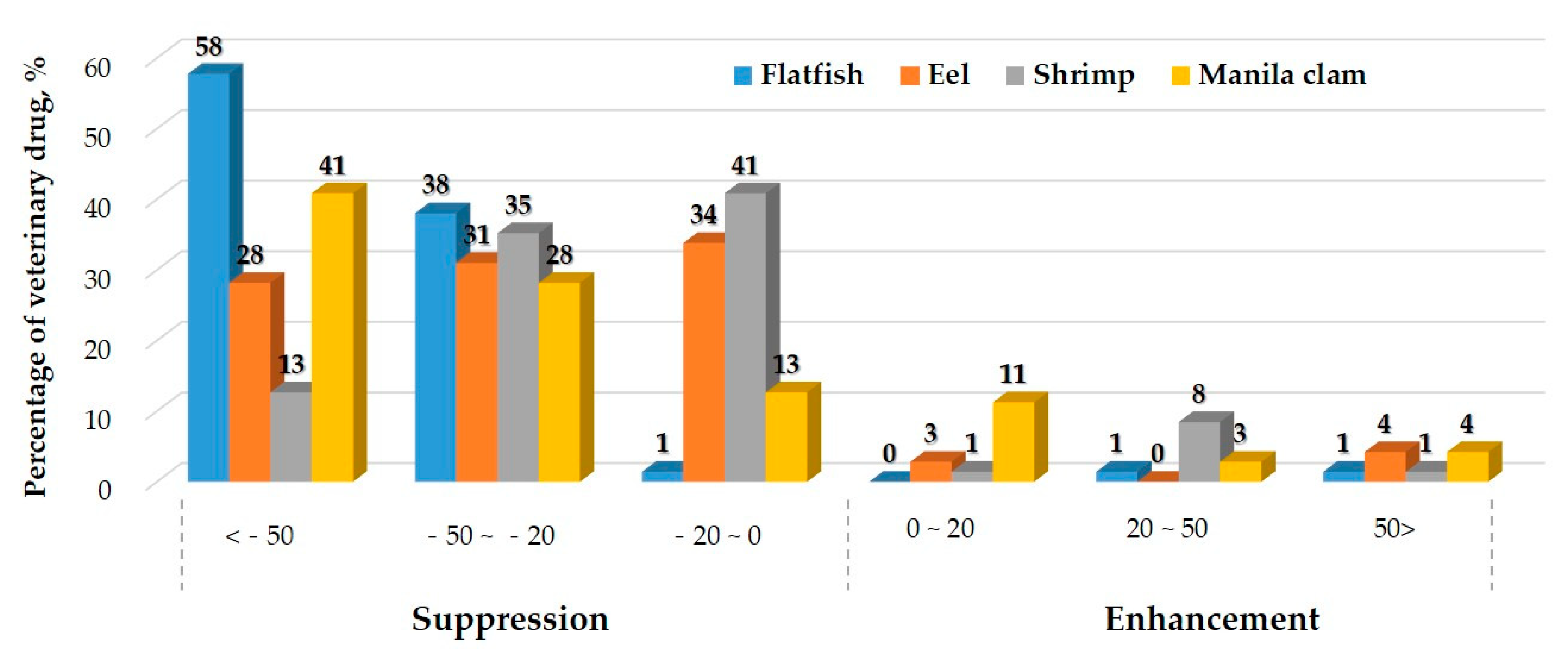
2.3.2. Matrix Effect
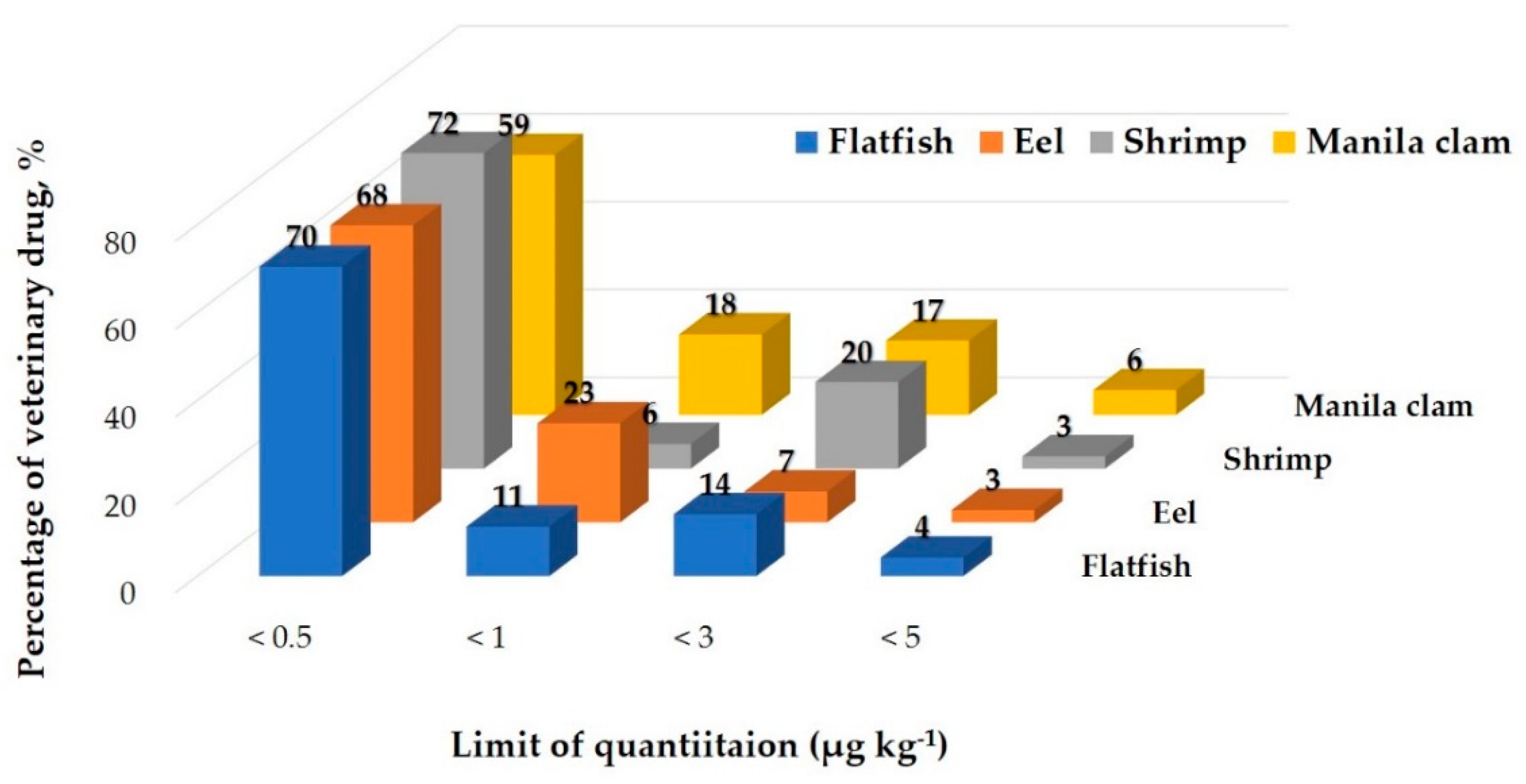
2.4. Application to Real Samples
3. Materials and Methods
3.1. Chemicals, Standards, and Stock Solutions
3.2. Sample Collection and Preparation
3.3. LC-MS/MS Conditions
3.4. Method Validation and Matrix Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Baiak, B.H.B.; Lehnen, C.R.; da Rocha, R.A. Anthelmintic resistance in cattle: A systematic review and meta-analysis. Livest. Sci. 2018, 217, 127–135. [Google Scholar] [CrossRef]
- Tuck, S.; Furey, A.; Danaher, M. Analysis of Anthelmintic and Anticoccidial Drug Residues in Animal-Derived Foods. In Chemical Analysis of Non-antimicrobial Veterinary Drug Residues in Food; Kay, J.F., MacNeil, J.D., Wang, J., Eds.; John Wiley & Sons, Inc.: Whitehouse, NJ, USA, 2016; pp. 245–309. [Google Scholar]
- Shao, Z.J. Aquaculture pharmaceuticals and biologicals: Current perspectives and future possibilities. Adv. Drug Deliv. Rev. 2001, 50, 229–243. [Google Scholar] [CrossRef]
- Stock, M.L.; Elazab, S.T.; Hsu, W.H. Review of triazine antiprotozoal drugs used in veterinary medicine. J. Vet. Pharmacol. Ther. 2018, 41, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Mundt, H.C.; Daugschies, A.; Uebe, F.; Rinke, M. Efficacy of toltrazuril against artificial infections with Eimeria bovis in calves. Parasitol Res. 2003, 90, S166–S167. [Google Scholar] [CrossRef]
- Gjerde, B.; Helle, O. Efficacy of toltrazuril in the prevention of coccidiosis in naturally infected lambs on pasture. Acta Vet. Scand. 1986, 27, 124–137. [Google Scholar] [CrossRef]
- Driesen, S.J.; Fahy, V.A.; Carland, P.G. The use of toltrazuril for the prevention of coccidiosis in piglets before weaning. Aust. Vet. J. 1995, 72, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Committee for veterinary medical products (CVMP), 1997. Metronidazole summary report EMEA/MRL/173/96-FINAL. European Medicines Agency. Retrieved 2009-12-11. Available online: https://www.ema.europa.eu/en/documents/mrl-report/metronidazole-summary-report-committee-veterinary-medicinal-products_en.pdf (accessed on 23 March 2021).
- Capitan-Vallvey, L.F.; Ariza, A.; Checa, R.; Navas, N. Liquid chromatography-mass spectrometry determination of six 5-nitroimidazoles in animal feedstuff. Chromatographia 2007, 65, 283–290. [Google Scholar] [CrossRef]
- Huet, A.C.; Mortier, L.; Daeseleire, E.; Fodey, T.; Elliott, C.; Delahaut, P. Development of an ELISA screening test for nitroimidazoles in egg and chicken muscle. Anal. Chim. Acta 2005, 534, 157–162. [Google Scholar] [CrossRef]
- European Commission Staff Working Document on the Implementation of National Residue Monitoring Plans in the Member States in 2004, Sanco/3400/2005, Annex 21–22. 2005. Available online: COM_SEC(2006)0224_EN.pdf (accessed on 23 March 2021).
- EUROPEAN COMMISSION. Commission Regulation (EC) No. 508/1999 of 4 March 1999 on laying down a community procedure for the establishment of maximum residue limits of veterinary medicinal products in foodstuffs of animal origin. Off. J. Eur. Union 1999, 60, 16. [Google Scholar]
- Ministry of Food and Drug Safety, South Korea. Korean Food Code. 2020. Available online: http://www.foodsafetykorea.go.kr/foodcode/01_02.jsp?idx=263 (accessed on 23 March 2021).
- Munoz, I.; Bueno, M.J.M.; Aguera, A.; Fernandez-Alba, A.R. Environmental and human health risk assessment of organic micro-pollutants occurring in a Spanish marine fish farm. Environ. Pollut. 2010, 158, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Beynon, S.A. Potential environmental consequences of administration of ectoparasiticides to sheep. Vet. Parasitol. 2012, 189, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Boxall, A.B.A.; Fogg, L.A.; Blackwell, P.A.; Blackwell, P.; Kay, P.; Pemberton, E.J.; Croxford, A. Veterinary medicines in the environment. Rev. Environ. Contam. Toxicol. 2004, 180, 1–91. [Google Scholar] [PubMed]
- Celiz, M.D.; Tso, J.; Aga, D.S. Pharmaceutical metabolites in the environment: Analytical challenges and ecological risks. Environ. Toxicol Chem 2009, 28, 2473–2484. [Google Scholar] [CrossRef]
- Kim, K.R.; Owens, G.; Kwon, S.I.; So, K.H.; Lee, D.B.; Ok, Y.S. Occurrence and environmental fate of veterinary antibiotics in the terrestrial environment. Water Air Soil Pollut 2011, 214, 163–174. [Google Scholar] [CrossRef]
- Boleda, M.R.; Galceran, M.T.; Ventura, F. Validation and uncertainty estimation of a multiresidue method for pharmaceuticals in surface and treated waters by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2013, 1286, 146–158. [Google Scholar] [CrossRef]
- Mainero, R.L.; Gentili, A.; Perez-Fernandez, V.; Tomai, P. Veterinary drugs residues: A review of the latest analytical research on sample preparation and LC-MS based methods. Food Addit. Contam. Part. A 2017, 34, 766–784. [Google Scholar] [CrossRef]
- Whelan, M.; Chirollo, C.; Furey, A.; Cortesi, M.L.; Anastasio, A.; Danaher, M. Investigation of the persistence of levamisole and oxyclozanide in milk and fate in cheese. J. Agric. Food Chem. 2010, 58, 12204–12209. [Google Scholar] [CrossRef]
- Kinsella, B.; Byrne, P.; Cantwell, H.; McCormack, M.; Furey, A.; Danaher, M. Determination of the new anthelmintic monepantel and its sulfone metabolite in milk and muscle using a UHPLC–MS/MS and QuEChERS method. J. Chromatogr. B 2011, 879, 3707–3713. [Google Scholar] [CrossRef]
- Hoff, R.B.; Molognoni, L.; Deolindo, C.T.P.; Vargas, M.O.; Kleemann, C.R.; Daguer, H. Determination of 62 veterinary drugs in feedingstuffs by novel pressurized liquid extraction methods and LC-MS/MS. J. Chromatogr. B 2020, 1152, 122232. [Google Scholar] [CrossRef]
- Sero, R.; Nunez, O.; Bosch, J.; Grases, J.M.; Rodriguez, P.; Moyano, E.; Galceran, M.T. Desorption electrospray ionization-high resolution mass spectrometry for the screening of veterinary drugs in cross-contaminated feedstuffs. Anal. Bioanal. Chem. 2015, 407, 7369–7378. [Google Scholar] [CrossRef] [Green Version]
- Dasenaki, M.E.; Bletsou, A.A.; Koulis, G.A.; Thomaidis, N.S. Qualitative multi residue screening method for 143 veterinary drugs and pharmaceuticals in milk and fish tissue using liquid chromatography quadrupole-time-of-flight mass spectrometry. J. Agric. Food Chem. 2015, 63, 4493–4508. [Google Scholar] [CrossRef] [PubMed]
- Maher, H.M.; Youssef, R.M.; Khalil, R.H.; El-Bahr, S.M. Simultaneous multiresidue determination of metronidazole and spiramycin in fish muscle using high performance liquid chromatography with UV detection. J. Chromatogr. B 2008, 876, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, B.; Lehotay, S.J.; Mastovska, K.; Lightfield, A.R.; Furey, A.; Danaher, M. New method for the analysis of flukicide and other anthelmintic residues in bovine milk and liver using liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2009, 637, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Cronly, M.; Behan, P.; Foley, B.; Malone, E.; Regan, L. Rapid confirmatory method for the determination of 11 nitroimidazoles in egg using liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2009, 1216, 8101–8109. [Google Scholar] [CrossRef] [PubMed]
- Frenich, A.G.; del Mar Aguilera-Luiz, M.; Vidal, J.L.M.; Romero-Gonzalez, R. Comparison of several extraction techniques for multiclass analysis of veterinary drugs in eggs using ultra-high pressure liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2010, 661, 150–160. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Quantification of anthelmintic drug residues in milk and muscle tissues by liquid chromatography coupled to Orbitrap and liquid chromatography coupled to tandem mass spectrometry. Talanta 2011, 85, 991–1000. [Google Scholar] [CrossRef]
- Mooney, D.; Coxon, C.; Richards, K.G.; Gill, L.; Mellander, P.E.; Danaher, M. Development and optimisation of a multiresidue method for the determination of 40 anthelmintic compounds in environmental water samples by solid phase extraction (SPE) with LC-MS/MS detection. Molecules 2019, 24, 1978. [Google Scholar] [CrossRef] [Green Version]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2015, 880, 103–121. [Google Scholar] [CrossRef]
- Chang, S. H.; Lai, Y.H.; Huang, C.N.; Peng, G.J.; Liao, C.D.; Kao, Y.M.; Wang, D.Y. Multi-residue analysis using liquid chromatography tandem mass spectrometry for detection of 20 coccidiostats in poultry, livestock, and aquatic tissues. J. Food Drug Anal. 2019, 27, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.; Moloney, M.; O’Mahony, J.; O’Kennedy, R.; Danaher, M. Determination of 20 coccidiostats in milk, duck muscle and non-avian muscle tissue using UHPLC-MS/MS. Food Addit. Contam. Part. A 2013, 30, 958–969. [Google Scholar] [CrossRef]
- Zrncic, M.; Gros, M.; Babic, S.; Kastelan-Macan, M.; Barcelo, D.; Petrovic, M. Analysis of anthelmintics in surface water by ultra high performance liquid chromatography coupled to quadrupole linear ion trap tandem mass spectrometry. Chemosphere 2014, 99, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.P.; Reyes, R.C.; Romero-Gonzalez, R.; Vidal, J.L.M.; Frenich, A.G. Multiresidue determination of veterinary drugs in aquaculture fish samples by ultra high performance liquid chromatography coupled to tandem mass spectrometry. J. Chromatogr. B 2012, 895, 39–47. [Google Scholar] [CrossRef]
- Yoo, K.H.; Park, D.H.; Abd El-Aty, A.M.; Kim, S.K.; Jung, H.N.; Jeong, D.H.; Shin, H.C. Development of an analytical method for multi-residue quantification of 18 anthelmintics in various animal-based food products using liquid chromatography-tandem mass spectrometry. J. Pharm. Anal. 2020, 11, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Afonsoa, I.; Loyo-Rosalesb, J.E.; de la Paz Aviles, M.; Rattner, B.A.; Rice, C.P. Determination of alkylphenol and alkylphenolethoxylates in biota by liquid chromatography with detection by tandem mass spectrometry and fluorescence spectroscopy. J. Chromatogr. A 2010, 1010, 25–35. [Google Scholar] [CrossRef]
- Whelan, M.; Kinsella, B.; Furey, A.; Moloney, M.; Cantwell, H.; Lehotay, S.J.; Danaher, M. Determination of anthelmintic drug residues in milk using ultra high performance liquid chromatography–tandem mass spectrometry with rapid polarity switching. J. Chromatogr. A 2010, 1217, 4612–4622. [Google Scholar] [CrossRef] [PubMed]
- Codex Alimentarius. Guidelines for the Design and Implementation of National Regulatory Food Safety Assurance Programme Associated with the Use of Veterinary Drugs in Food Producing Animals CAC/GL 71. 2009. Available online: http://www.fao.org/input/download/standards/11252/CXG_071e_2014.pdf (accessed on 23 March 2021).
- Whelan, M.; O’Mahony, J.; Moloney, M.; Cooper, K.M.; Furey, A.; Kennedy, D.G.; Danaher, M. Maximum residue level validation of triclabendazole marker residues in bovine liver, muscle and milk matrices by ultra high pressure liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2013, 1275, 41–47. [Google Scholar] [CrossRef]
- Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M.C. Signal suppression/enhancement in high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. [Google Scholar] [CrossRef]
- Ferrer, C.; Lozano, A.; Aguera, A.; Giron, A.J.; Fernández-Alba, A.R. Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables. J. Chromatogr. A 2011, 1218, 7634–7639. [Google Scholar] [CrossRef]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An overview of matrix effects in liquid chromatography–mass spectrometry. Mass Spectrom Rev. 2011, 30, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, H.; Reemtsma, T.; Alder, L. Compensation of matrix effects by post column infusion of a monitor substance in multiresidue analysis with LC− MS/MS. Anal. Chem. 2009, 81, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Stuber, M.; Reemtsma, T. Evaluation of three calibration methods to compensate matrix effects in environmental analysis with LC-ESI-MS. Anal. Bioanal. Chem. 2004, 378, 910–916. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Recovery | Number of Veterinary Drugs (Percentage, %) | |||
|---|---|---|---|---|
| Flatfish | Eel | Shrimp | Manila Clam | |
| L1 | (CV 2.3%~28.4%) | (CV 2.8%~30.2%) | (CV 0.8%~26.6%) | (CV 1.7%~29.7%) |
| 60% to 80% | 3(4.2%) | 7(9.9%) | 11(15.5%) | 5(7.0%) |
| 80% to 100% | 38(53.5%) | 35(49.3%) | 35(49.3%) | 30(42.3%) |
| 100% to 120% | 30(42.3%) | 29(40.8%) | 25(35.2%) | 36(50.7%) |
| L2 | (CV 1.7%~23.1%) | (CV 1.5%~24.6%) | (CV 1.1%~14.2%) | (CV 2.2%~27.6%) |
| 60% to 80% | 1(1.4%) | 1(1.4%) | 0(0.0%) | 2(2.8%) |
| 80% to 100% | 24(33.8%) | 29(40.8%) | 38(53.5%) | 31(43.7%) |
| 100% to 120% | 46(64.8%) | 41(57.7%) | 33(46.5%) | 38(53.5%) |
| L3 | (CV 0.9%~18.8%) | (CV 1.5%~20.4%) | (CV 1.0%~17.5%) | (CV 1.8%~22.2%) |
| 60% to 80% | 1(1.4%) | 1(1.4%) | 0(0.0%) | 1(1.4%) |
| 80% to 100% | 33(46.5%) | 37(52.1%) | 44(62.0%) | 11(15.5%) |
| 100% to 120% | 37(52.1%) | 33(46.5%) | 27(38.0%) | 59(83.1%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.; Park, S.; Park, H.; Choi, J.; Yoon, H.J.; Kim, J.-H. Determination of Anthelmintic and Antiprotozoal Drug Residues in Fish Using Liquid Chromatography-Tandem Mass Spectrometry. Molecules 2021, 26, 2575. https://doi.org/10.3390/molecules26092575
Kim E, Park S, Park H, Choi J, Yoon HJ, Kim J-H. Determination of Anthelmintic and Antiprotozoal Drug Residues in Fish Using Liquid Chromatography-Tandem Mass Spectrometry. Molecules. 2021; 26(9):2575. https://doi.org/10.3390/molecules26092575
Chicago/Turabian StyleKim, Eunjung, Sihyun Park, Hyunjin Park, Jangduck Choi, Hae Jung Yoon, and Jeong-Han Kim. 2021. "Determination of Anthelmintic and Antiprotozoal Drug Residues in Fish Using Liquid Chromatography-Tandem Mass Spectrometry" Molecules 26, no. 9: 2575. https://doi.org/10.3390/molecules26092575
APA StyleKim, E., Park, S., Park, H., Choi, J., Yoon, H. J., & Kim, J. -H. (2021). Determination of Anthelmintic and Antiprotozoal Drug Residues in Fish Using Liquid Chromatography-Tandem Mass Spectrometry. Molecules, 26(9), 2575. https://doi.org/10.3390/molecules26092575