
Deglycosylated Azithromycin Attenuates Bleomycin-Induced Pulmonary Fibrosis via the TGF-β1 Signaling Pathway

 , and
, and
Abstract
:1. Introduction
2. Results
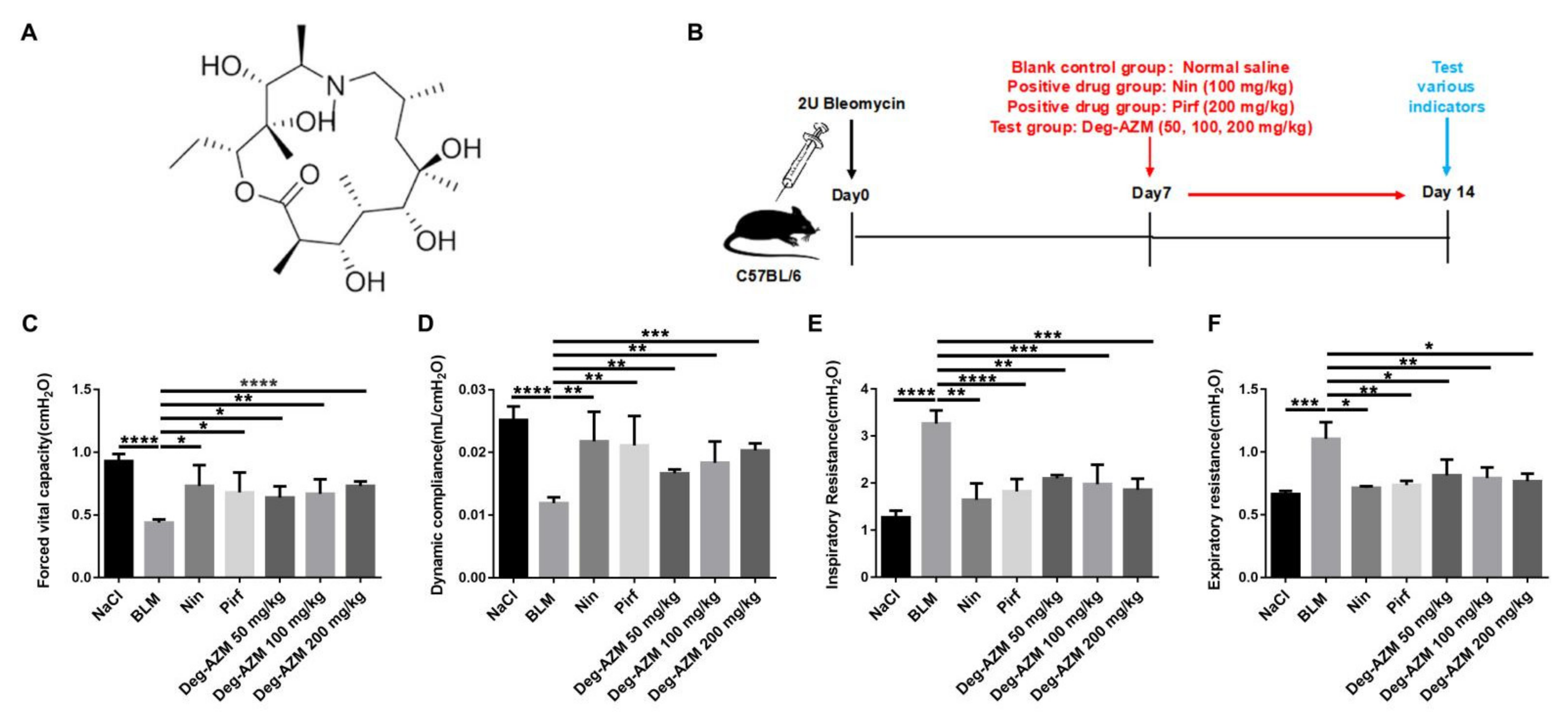
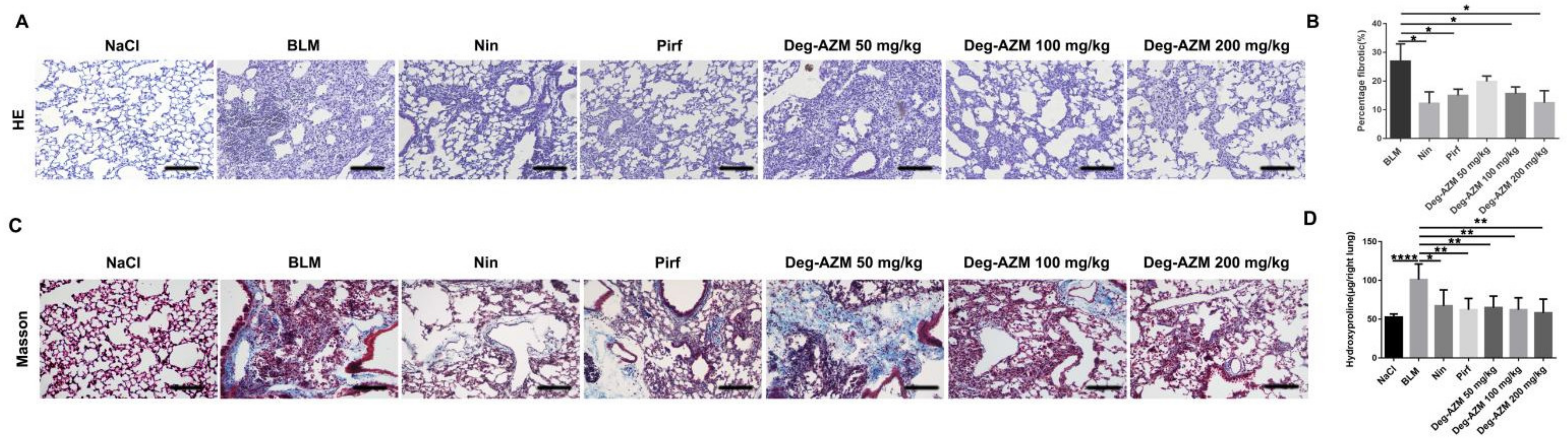
2.1. Deglycosylated Azithromycin Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice
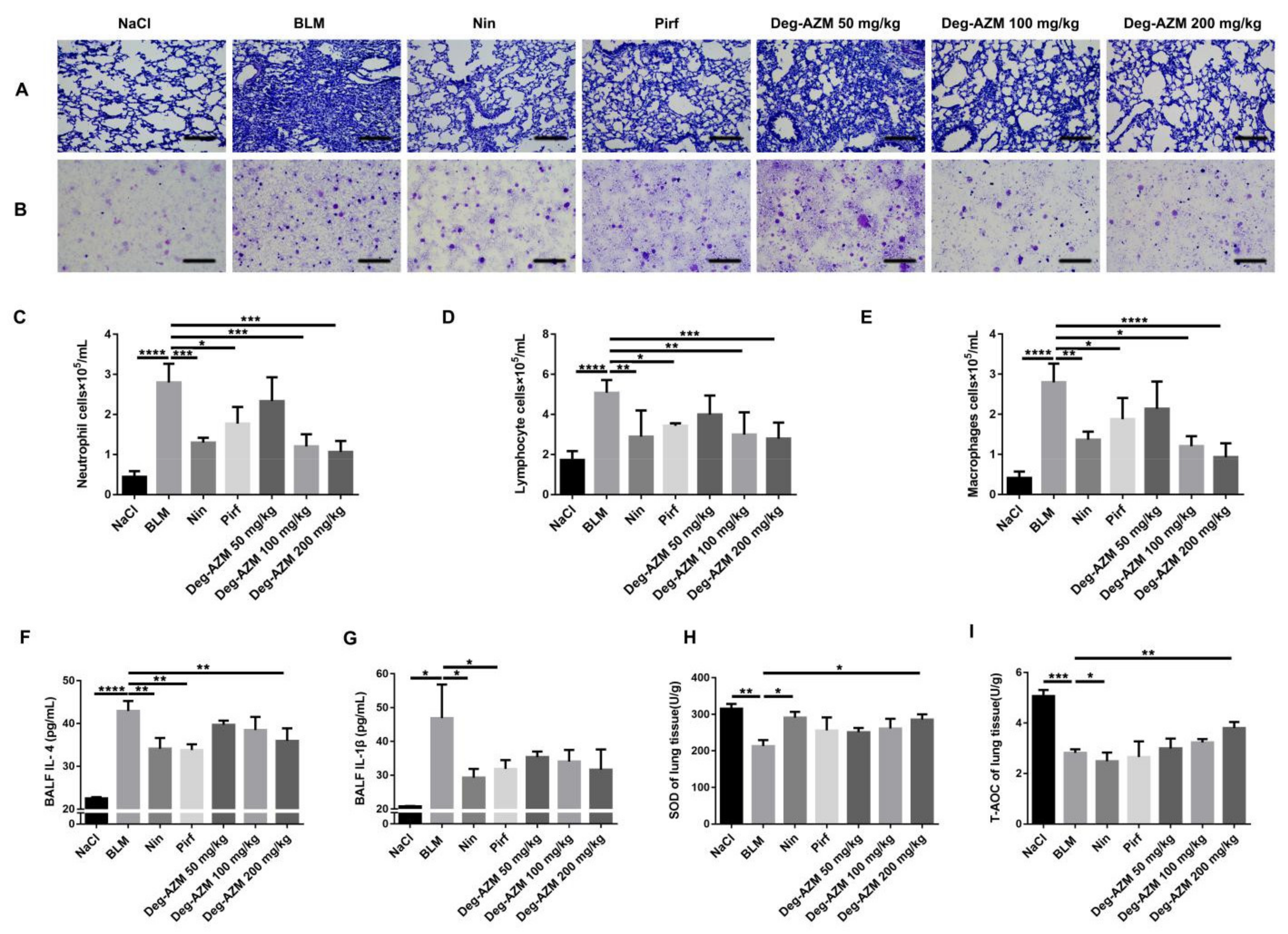
2.2. Deglycosylated Azithromycin Suppresses the Inflammatory Response and Oxidative Stress in Vivo
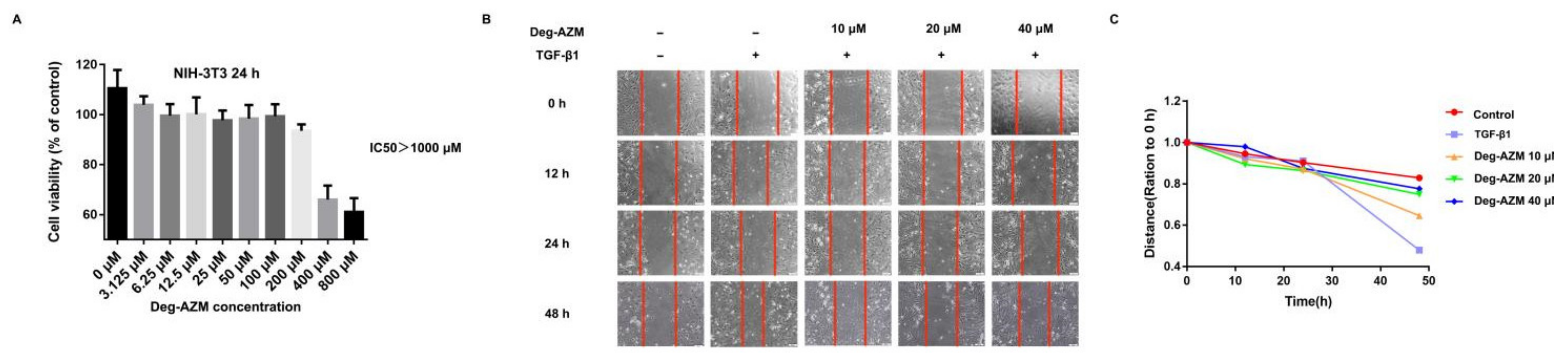
2.3. Deglycosylated Azithromycin Inhibits TGF-β1-Induced Fibroblast Proliferation and Migration
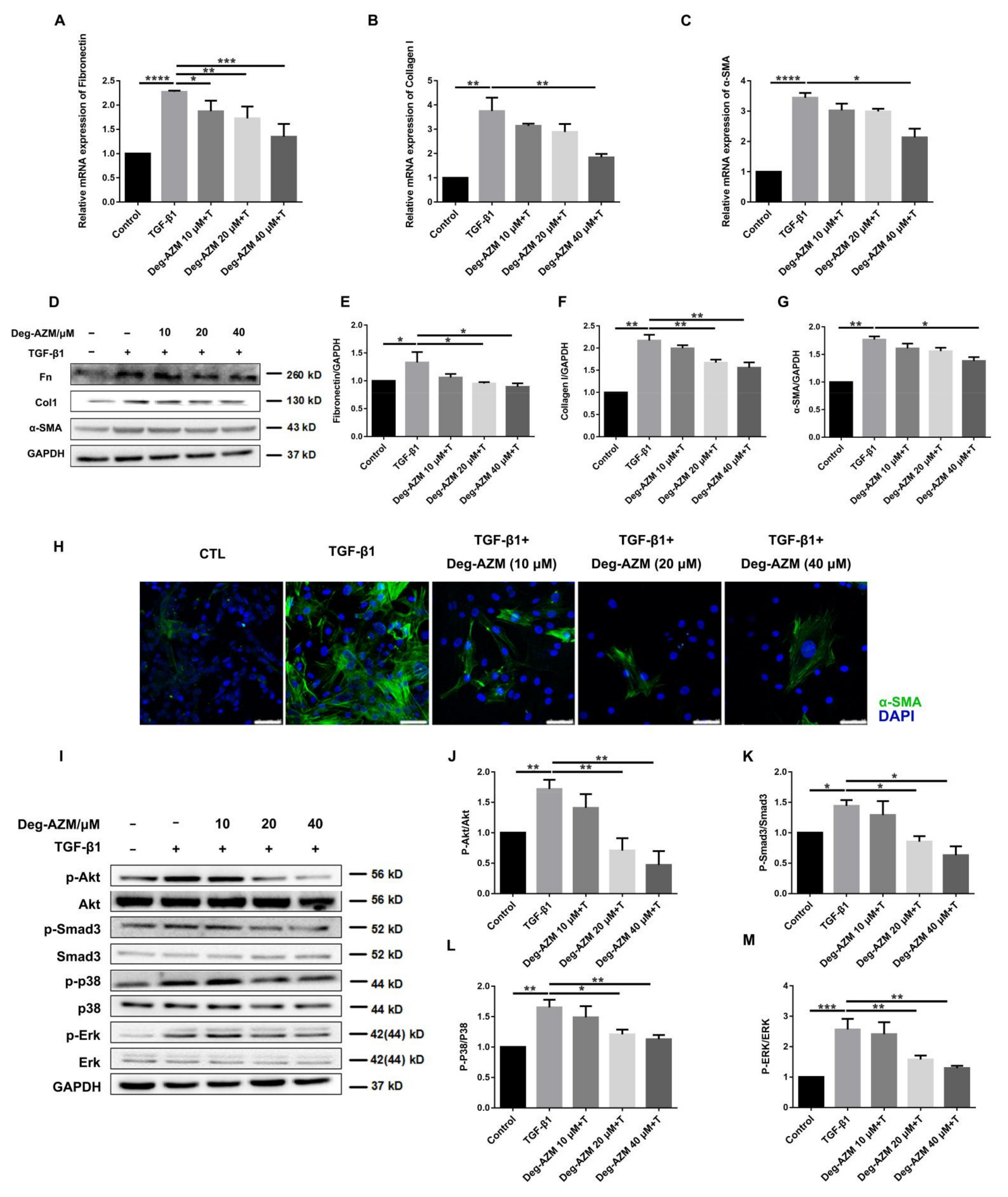
2.4. Deglycosylated Azithromycin Attenuates TGF-β1-Induced Myofibroblast Activation
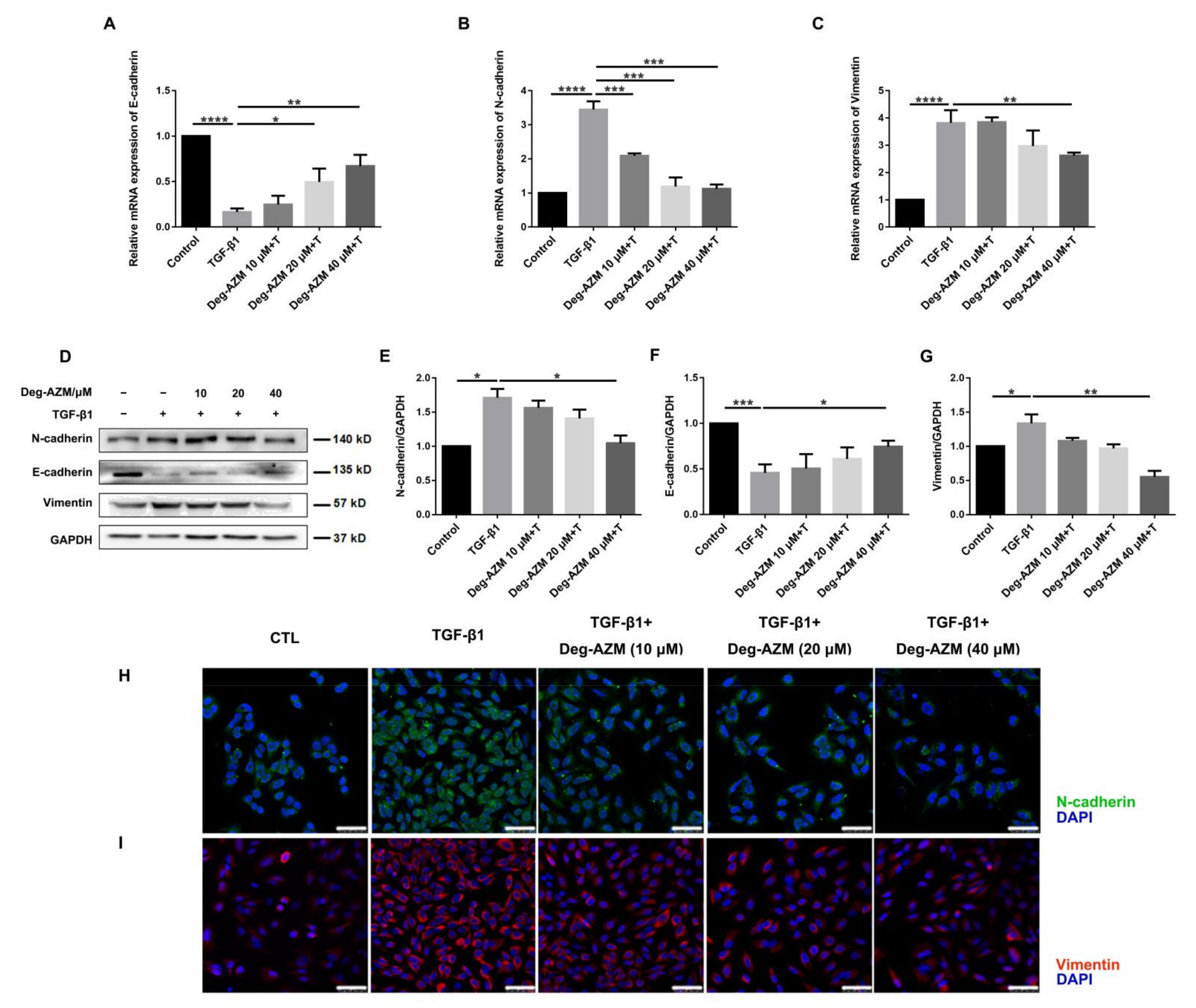
2.5. Deglycosylated Azithromycin Inhibits TGF-β1-Induced EMT in Epithelial Cells
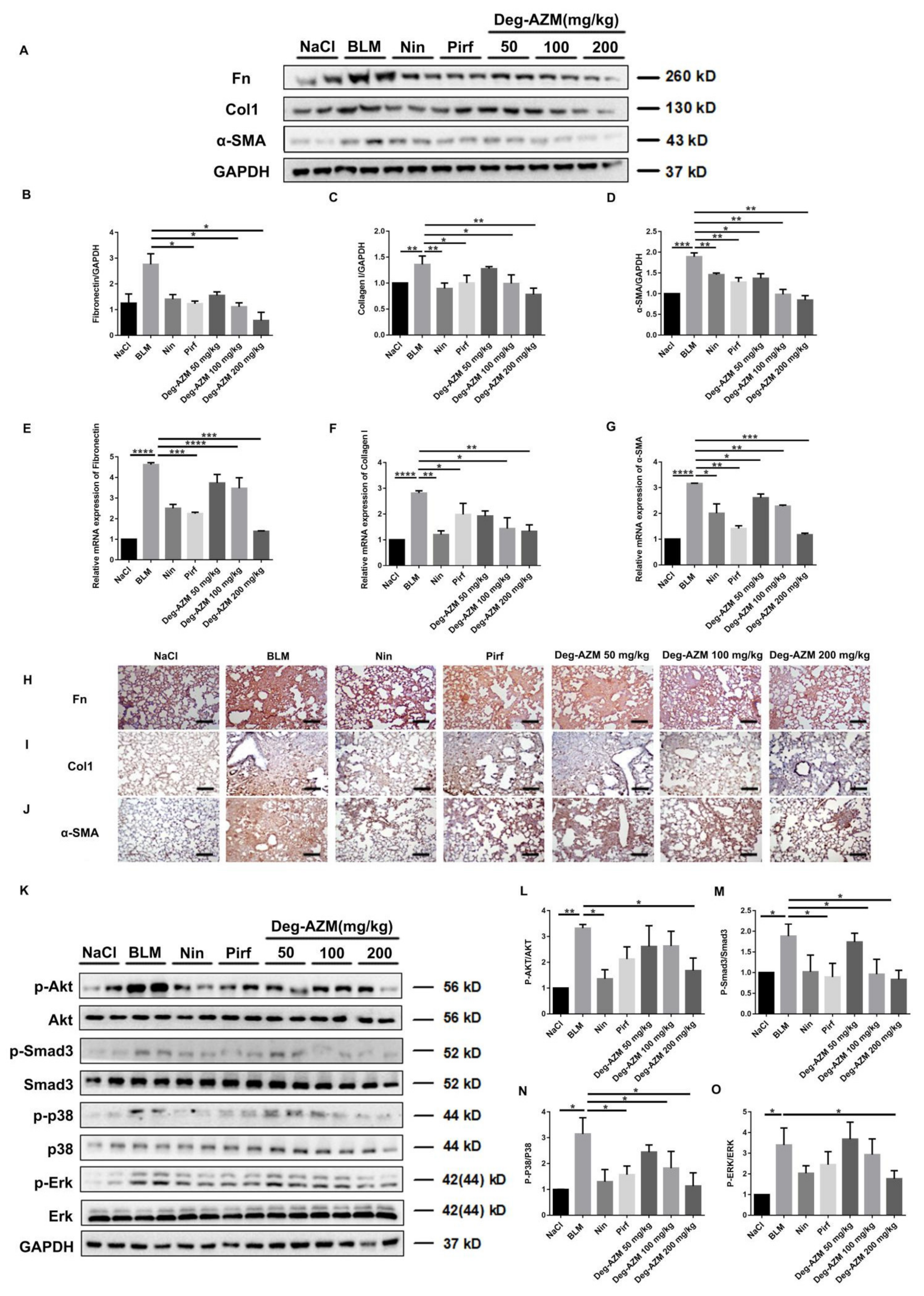
2.6. Deglycosylated Azithromycin Inhibits the Fibrogenic Activation of Pulmonary Fibroblasts in Vivo
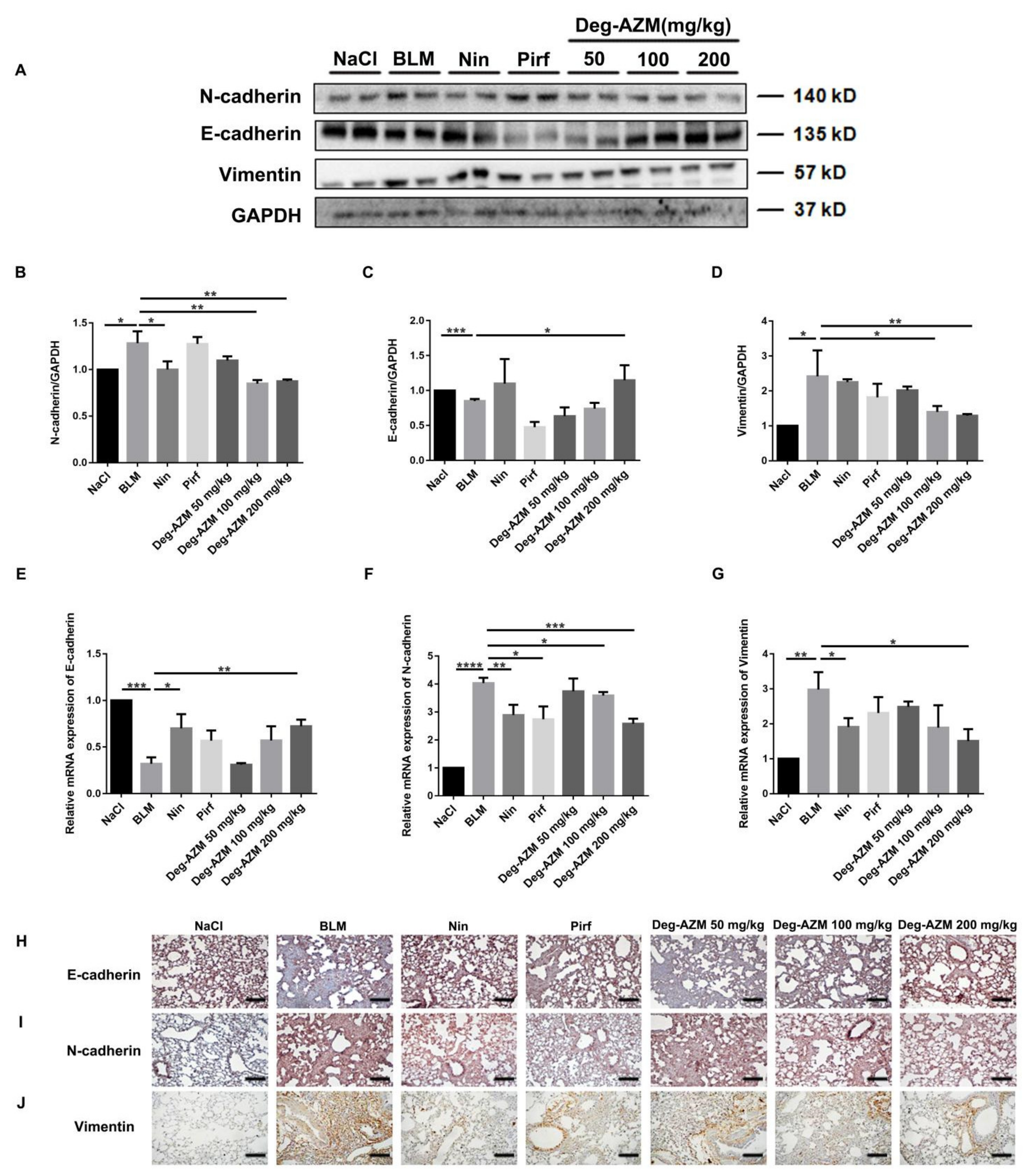
2.7. Deglycosylated Azithromycin Inhibits the EMT Phenotype in Vivo
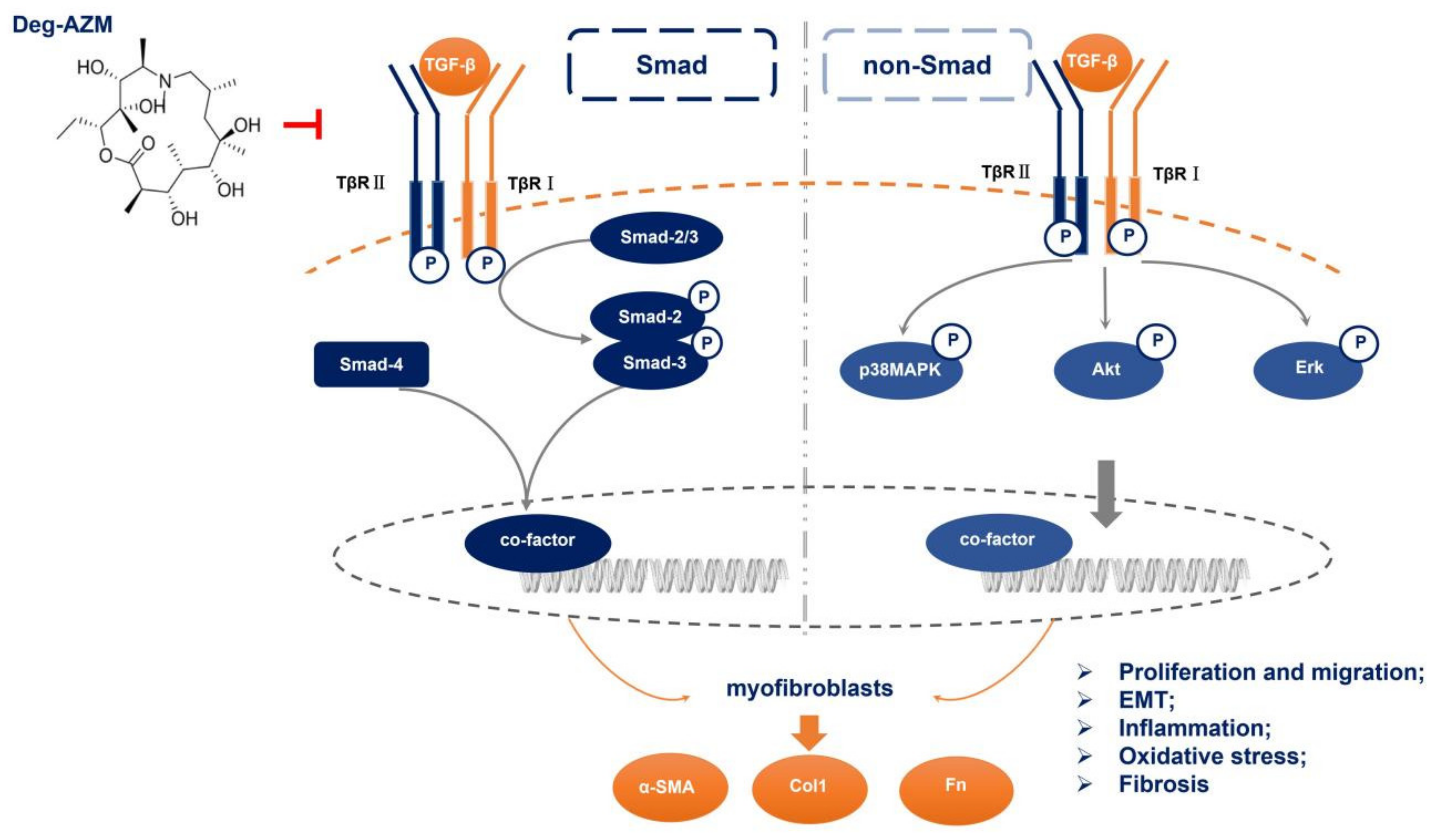
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Animals
4.4. Bleomycin Administration
4.5. Pulmonary Function Testing
4.6. Hydroxyproline Measurement
4.7. Histological Examination
4.8. Immunohistochemistry Staining
4.9. ELISA for the Detection of Inflammatory Factors
4.10. Measurement of Oxidative Stress
4.11. Bronchoalveolar Lavage
4.12. Quantitative Real-Time PCR (qRT-PCR)
4.13. Western Blotting Analysis
4.14. Immunofluorescence Staining
4.15. Cell Viability Analysis
4.16. Wound-Healing Assay
4.17. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Jee, A.S.; Sahhar, J.; Youssef, P.; Bleasel, J.; Adelstein, S.; Nguyen, M.; Corte, T.J. Review: Serum biomarkers in idiopathic pulmonary fibrosis and systemic sclerosis associated interstitial lung disease – frontiers and horizons. Pharmacol. Ther. 2019, 202, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Thannickal, V.J. The Aging Lung and Idiopathic Pulmonary Fibrosis. Am. J. Med Sci. 2019, 357, 384–389. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and man-agement. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Mamazhakypov, A.; Schermuly, R.T.; Schaefer, L.; Wygrecka, M. Lipids - two sides of the same coin in lung fibrosis. Cell. Signal. 2019, 60, 65–80. [Google Scholar] [CrossRef]
- Kinoshita, T.; Goto, T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int. J. Mol. Sci. 2019, 20, 1461. [Google Scholar] [CrossRef] [Green Version]
- Bisping, G.; Kropff, M.; Wenning, D.; Dreyer, B.; Bessonov, S.; Hilberg, F.; Roth, G.J.; Munzert, G.; Stefanic, M.; Stelljes, M.; et al. Targeting receptor kinases by a novel indolinone derivative in multiple myeloma: Abrogation of stroma-derived interleukin-6 secretion and induction of apoptosis in cytogenetically defined subgroups. Blood 2006, 107, 2079–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Xu, W.; Chen, H.; Warburton, D.; Dong, R.; Qian, B.; Selman, M.; Gauldie, J.; Kolb, M.; Shi, W. A novel profibrotic mechanism mediated by TGFbeta-stimulated collagen prolyl hydroxylase expression in fibrotic lung mesenchymal cells. J. Pathol. 2015, 236, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17. [Google Scholar] [CrossRef]
- Salton, F.; Volpe, M.; Confalonieri, M. Epithelial–Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Med. 2019, 55, 83. [Google Scholar] [CrossRef] [Green Version]
- Corvol, H.; Flamein, F.; Epaud, R.; Clement, A.; Guillot, L. Lung alveolar epithelium and interstitial lung disease. Int. J. Biochem. Cell Biol. 2009, 41, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Cheng, D.S.; Degryse, A.L.; Zoz, D.F.; Abdolrasulnia, R.; Lawson, W.E.; Blackwell, T.S. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J. Biol. Chem. 2011, 286, 30972–30980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L.M. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharaj, S.; Shimbori, C.; Kolb, M. Fibrocytes in pulmonary fibrosis: A brief synopsis. Eur. Respir. Rev. 2013, 22, 552–557. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, W.A.; Willems, S.; Vos, R.; Vanaudenaerde, B.M.; De Vleeschauwer, S.I.; Rinaldi, M.; Vanhooren, H.M.; Geudens, N.; Verleden, S.E.; Demedts, M.G.; et al. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp. Lung Res. 2010, 36, 602–614. [Google Scholar] [CrossRef]
- Macaluso, C.; Furcada, J.M.; Alzaher, O.; Chaube, R.; Chua, F.; Wells, A.U.; Maher, T.M.; George, P.M.; Renzoni, E.A.; Molyneaux, P.L. The potential impact of azithromycin in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53, 1800628. [Google Scholar] [CrossRef]
- Zhong, W.; Sun, B.; Ruan, H.; Yang, G.; Qian, B.; Cao, H.; He, L.; Fan, Y.; Roberts, A.G.; Liu, X.; et al. Deglycosylated Azithromycin Targets Transgelin to Enhance Intestinal Smooth Muscle Function. iScience 2020, 23, 101464. [Google Scholar] [CrossRef] [PubMed]
- Heukels, P.; Moor, C.; von der Thüsen, J.; Wijsenbeek, M.; Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-L.; Xing, R.-G.; Chen, L.; Liu, C.-R.; Miao, Z.-G. PI3K/Akt signaling is involved in the pathogenesis of bleomycin-induced pulmonary fibrosis via regulation of epithelial-mesenchymal transition. Mol. Med. Rep. 2016, 14, 5699–5706. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Agostini, C.; Antoniou, K.M.; Bouros, D.; Chambers, R.C.; Cottin, V.; Egan, J.J.; Lambrecht, B.N.; Lories, R.; Parfrey, H.; et al. The pathogenesis of pulmonary fibrosis: A moving target. Eur. Respir. J. 2012, 41, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Sgalla, G.; Biffi, A.; Richeldi, L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology 2015, 21, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, M.; Kim, Y.O.; Wagner, W.L.; Schuppan, D.; Valenzuela, C.D.; Mentzer, S.J.; Kreuz, S.; Stiller, D.; Wollin, L.; Konerding, M.A. Effects of nintedanib on the microvascular architecture in a lung fibrosis model. Angiogenesis 2017, 20, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Ichikado, K.; Yasuda, Y.; Anan, K.; Suga, M. Azithromycin for idiopathic acute exacerbation of idiopathic pulmonary fibrosis: A retrospective single-center study. BMC Pulm. Med. 2017, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.G.; Moore, B.B.; Chambers, R.C.; Eickelberg, O.; Königshoff, M.; Kolb, M.; Laurent, G.J.; Nanthakumar, C.B.; Olman, M.A.; Pardo, A.; et al. An Official American Thoracic Society Workshop Report: Use of Animal Models for the Preclinical Assessment of Potential Therapies for Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Barlo, N.P.; van Moorsel, C.H.; Korthagen, N.M.; Heron, M.; Rijkers, G.T.; Ruven, H.J.; van den Bosch, J.M.; Grutters, J.C. Genetic variability in the IL1RN gene and the balance between interleukin (IL)-1 receptor agonist and IL-1beta in idiopathic pulmonary fibrosis. Clin. Exp. Immunol. 2011, 166, 346–351. [Google Scholar] [CrossRef]
- Vu, T.N.; Chen, X.; Foda, H.D.; Smaldone, G.C.; Hasaneen, N.A. Interferon-gamma enhances the antifibrotic effects of pirfenidone by attenuating IPF lung fibroblast activation and differentiation. Respir. Res. 2019, 20, 206. [Google Scholar] [CrossRef] [Green Version]
- Geudens, N.; Timmermans, L.; Vanhooren, H.; Vanaudenaerde, B.M.; Vos, R.; Van De Wauwer, C.; Verleden, G.M.; Verbeken, E.; Lerut, T.; Van Raemdonck, D.E. Azithromycin reduces airway inflammation in a murine model of lung ischaemia reper-fusion injury. Transpl. Int. 2008, 21, 688–695. [Google Scholar] [CrossRef]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S.; et al. Contribution of Epithelial-derived Fibroblasts to Bleomycin-induced Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- John, A.E.; Graves, R.H.; Pun, K.T.; Vitulli, G.; Forty, E.J.; Mercer, P.F.; Morrell, J.L.; Barrett, J.W.; Rogers, R.F.; Hafeji, M.; et al. Translational pharmacology of an inhaled small molecule alphavbeta6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat. Commun. 2020, 11, 4659. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Guo, J.; Fang, Y.; Jiang, F.; Li, L.; Zhou, H.; Xu, X.; Ning, W. Neohesperidin inhibits TGF-β1/Smad3 signaling and alleviates bleomycin-induced pulmonary fibrosis in mice. Eur. J. Pharmacol. 2019, 864, 172712. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Geng, Y.; Li, L.; Li, X.; Yan, X.; Fang, Y.; Li, X.; Dong, S.; Liu, X.; Yang, X.; et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J. Exp. Med. 2015, 212, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Hodge, J.; Lu, B.; Zhu, Z.; Yu, S.; Fan, J.; Gao, Y.; Yin, Z.; Homer, R.; et al. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J. Clin. Investig. 2004, 114, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Bi, Z.; Liu, S.; Gao, S.; Cui, Y.; Huang, K.; Huang, M.; Mao, J.; Li, L.; Gao, J.; et al. Antifibrotic Mechanism of Cinobufagin in Bleomycin-Induced Pulmonary Fibrosis in Mice. Front. Pharmacol. 2019, 10, 1021. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence (5′–3′) | Reverse Primer Sequence (3′–5′) |
|---|---|---|
| Mouse GAPDH | AGGTCGGTGTGAACGGATTTG | GGGGTCGTTGATGGCAACA |
| Mouse α-SMA | GTCCCAGACATCAGGGAGTAA | GTCCCAGACATCAGGGAGTAA |
| Mouse Fibronectin | TCGGATACTTCAGCGTCAGGA | TCGGATACTTCAGCGTCAGGA |
| Mouse Collagen I | ATGTGGACCCCTCCTGATAGT | ATGTGGACCCCTCCTGATAGT |
| Mouse E-cadherin | CAGCCTTCTTTTCGGAAGACT | GGTAGACAGCTCCCTATGACTG |
| Mouse N-cadherin | CTCCAACGGGCATCTTCATTAT | CAAGTGAAACCGGGCTATCAG |
| Mouse Vimentin | GCTGCGAGAGAAATTGCAGGA | CCACTTTCCGTTCAAGGTCAAG |
| Human GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
| Human E-cadherin | ATTTTTCCCTCGACACCCGAT | TCCCAGGCGTAGACCAAGA |
| Human N-cadherin | TTTGATGGAGGTCTCCTAACACC | ACGTTTAACACGTTGGAAATGTG |
| Human Vimentin | AGTCCACTGAGTACCGGAGAC | CATTTCACGCATCTGGCGTTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, H.; Gao, S.; Li, S.; Luan, J.; Jiang, Q.; Li, X.; Yin, H.; Zhou, H.; Yang, C. Deglycosylated Azithromycin Attenuates Bleomycin-Induced Pulmonary Fibrosis via the TGF-β1 Signaling Pathway. Molecules 2021, 26, 2820. https://doi.org/10.3390/molecules26092820
Ruan H, Gao S, Li S, Luan J, Jiang Q, Li X, Yin H, Zhou H, Yang C. Deglycosylated Azithromycin Attenuates Bleomycin-Induced Pulmonary Fibrosis via the TGF-β1 Signaling Pathway. Molecules. 2021; 26(9):2820. https://doi.org/10.3390/molecules26092820
Chicago/Turabian StyleRuan, Hao, Shaoyan Gao, Shuangling Li, Jiaoyan Luan, Qiuyan Jiang, Xiaohe Li, Huijun Yin, Honggang Zhou, and Cheng Yang. 2021. "Deglycosylated Azithromycin Attenuates Bleomycin-Induced Pulmonary Fibrosis via the TGF-β1 Signaling Pathway" Molecules 26, no. 9: 2820. https://doi.org/10.3390/molecules26092820
APA StyleRuan, H., Gao, S., Li, S., Luan, J., Jiang, Q., Li, X., Yin, H., Zhou, H., & Yang, C. (2021). Deglycosylated Azithromycin Attenuates Bleomycin-Induced Pulmonary Fibrosis via the TGF-β1 Signaling Pathway. Molecules, 26(9), 2820. https://doi.org/10.3390/molecules26092820